通过色谱柱缩放和方法优化提高寡核苷酸的纯化效率
仅供研究使用,不适用于诊断。
摘要
多年来,小分子和蛋白质类治疗药物一直在制药行业中占主导地位。然而,这些药物模式在治疗靶点和治疗方法数量上存在局限性。合成寡核苷酸可考虑作为替代方案,尤其是在涉及遗传信息转录、剪接或翻译的应用领域。治疗性寡核苷酸是一类小型合成核酸聚合物,长度在20~30 mer之间,可用于调控基因表达。在药物开发过程中,从早期发现到后期开发,都需要纯化程度足够高的药物来支持一系列不同的研究。此外,聚合酶链式反应和定量基因组学方法都依赖寡核苷酸作为分子工具。随着色谱创新技术的不断涌现,纯化技术不断发展,纯化方案也不断改进。本研究提出了一种经济有效、高通量的系统化方法,用于20 mer寡核苷酸的分析级分离和制备级纯化。
优势
- 危害性更低、更经济实惠的流动相添加剂,在分馏后可轻松去除
- 将分析型UPLC分离色谱柱放大为大孔径纯化柱
- 采用批量测试和选定的寡核苷酸BEH™ C18固定相,确保分离和纯化性能的重现性
- 优化色谱方法参数,实现高效分离和纯化
- 优化制备型色谱柱的柱上载样量和馏分收集,尽可能提高纯度和回收率
简介
合成寡核苷酸的高效纯化通常是一项艰巨的任务。迄今为止,色谱工作者在选择合适的色谱柱、选择流动相和优化梯度条件方面一直面临着挑战。许多文献介绍了分析和表征寡核苷酸使用的分离方法1~3。分析方法通常通过使用高pH值和高温进行优化。选择固定相时必须考虑这些色谱分析需求。许多科学家选择亚乙基桥杂化硅胶(BEH) C18吸附剂用于此目的。Waters BEH C18吸附剂色谱柱提供有分析级和制备级尺寸,称为XBridge C18色谱柱。此外,沃特世还使用寡核苷酸分离方法对BEH C18颗粒进行批量测试和选择,进一步生产出ACQUITY™和XBridge™ BEH C18寡核苷酸分析专用柱。
寡核苷酸的液相色谱法主要采用反相离子对(RP-IP)分离,在流动相中使用烷基胺添加剂,并使用乙酸或六氟异丙醇(HFIP)进行缓冲。三乙胺(TEA)/HFIP水相流动相是寡核苷酸LC MS分析的金标准。使用这类流动相可获得优异的峰形,使用HFIP代替乙酸盐可减少离子抑制效应,从而使洗脱的寡核苷酸更容易被质谱检测到。
将寡核苷酸分离方法放大到制备级规模的一个挑战在于,HFIP的成本高且具有危险性。实验室可以选择替代色谱条件来进行常规制备规模的分离,从而降低成本并减少对HFIP的接触。其次,小粒径吸附剂通常会产生高压,不适用于制备型分离。解决这一问题的一个理想方案是降低流速并使用填充全多孔2.5 µm颗粒的色谱柱。沃特世提供用于实验室规模制备型纯化的色谱柱,内径(ID)从4.6 mm、10 mm到50 mm不等。用于制备型色谱柱的最佳柱床密度(OBD™)填充技术通过提高色谱柱的机械强度、重现性和柱效,延长了色谱柱的使用寿命,从而增强了高通量纯化的效果1。
在色谱分离中,使用基于乙酸三乙铵(TEAA)或乙酸己基铵(HAA)的流动相可以替代HFIP2。 TEAA和HAA均为挥发性物质,收集到的寡核苷酸馏分经过蒸发或冻干处理可将其去除。研究表明,增加碳链长度的各种替代烷基胺均可用作离子对试剂来分离寡核苷酸2。 烷基胺的选择会受到待纯化寡核苷酸性质的影响。但是,碳链长度的增加通常会降低挥发性,从而增加蒸发或冻干所需的时间和能耗成本。采用挥发性流动相和添加剂的方法对于降低成本、确保实验室安全以及限制纯化或纯化后步骤的数量至关重要。
本应用纪要展示了寡核苷酸纯化方法的放大过程,从基于TEA/HFIP流动相的分析型UPLC ™分离开始,到使用无HFIP的大孔径HPLC纯化方法为止。
实验
20 mer寡核苷酸模型混合物的LC分析
样品前处理
粗制的20 mer 5’-G-C-C-T-C-A-G-T-C-T-G-C-T-T-C-C-A-C-C-T-3购自Oligo Factory(美国马萨诸塞州霍利斯顿),在本研究中用作探针寡核苷酸化合物。将粗制混合物复溶于10 mM NH4OAc中,配制成总浓度为10 µM (59 µg/mL)的样品溶剂。
流动相制备
制备8.6/100 mM TEA/HFIP的混合物作为流动相A。使用MeOH作为流动相B。在其他实验中,制备100 mM TEAA (pH 7.0)用作流动相A,并使用乙腈作为流动相B。
色谱柱
本研究使用XBridge BEH C18 130 Å 2.5 µm寡核苷酸分析专用柱。该色谱柱包含经过专门测试和批次选择的BEH C18固定相,确保色谱柱在寡核苷酸分离中的性能。QC测试在美国马萨诸塞州陶顿进行,确保只有在离子对条件下能够产生尖锐、对称峰的固定相批次才会被用于寡核苷酸分析色谱柱。色谱工作者在寻求和保持重现性高的方法及纯化技术时是一个重要的考虑因素。
初始分析方法
|
流动相A: |
8.6/100 mM TEA/HFIP |
|
流动相B: |
MeOH |
|
色谱柱: |
ACQUITY UPLC BEH C18, 2.1 × 100 mm, 1.7 μm寡核苷酸分析专用柱(P/N:186003950) |
|
流速: |
0.4 mL/min梯度 |
|
柱温: |
60 ℃ |
|
稀释剂: |
10 mM NH4OAc水溶液 |
|
样品浓度: |
10 μM (59 μg/mL) |
|
进样体积: |
2 µL(柱上进样量120 ng) |
|
梯度: |
5%流动相B保持0.25 min,10 min内从5%增加到15%,0.25 min内从15%增加到95%,95%保持1 min,在0.1 min内从95%降到5%,5%保持3.4 min。 |
优化的TEAA分析方法
|
流动相A: |
100 mM TEAA,pH 7.0 |
|
流动相B: |
乙腈 |
|
色谱柱: |
XBridge BEH C18 130 Å寡核苷酸分析专用柱, 2.5 µm, 4.6 × 50 mm(P/N:186003953) |
|
流速: |
0.8 mL/min梯度 |
|
柱温: |
25 °C |
|
稀释剂: |
100 mM TEAA,pH 7.0 |
|
样品浓度: |
1 mg/mL |
|
进样体积: |
10 µL(柱上进样量10 µg) |
|
梯度: |
0.5 min内从1%增加至7.8%,在10 min内从7.8%增加至9.8%,在0.1 min内从9.8%增加至19%,保持19% 1.4 min,在0.1 min内从19%降至1%,保持1% 4.9 min |
优化的制备型TEAA方法
|
流动相A: |
100 mM TEAA,pH 7.0 |
|
流动相B: |
乙腈 |
|
色谱柱: |
XBridge BEH C18, 130 Å OBD, 30 × 50 mm, 2.5 µm寡核苷酸专用制备柱(P/N:186008963) |
|
流速: |
25 mL/min梯度 40 mL/min清洗和平衡 |
|
柱温: |
25 °C |
|
稀释剂: |
100 mM TEAA,pH 7.0 |
|
样品浓度: |
10 mg/mL |
|
进样体积: |
42 μL,然后变化 |
|
梯度: |
0.2 min内从1%增加至7.8%,在12 min内从7.8%增加至9.8%,在0.1 min内从9.8%增加至19%,保持19% 0.9 min,在0.1 min内从19%降至1%,保持1% 2 min |
结果与讨论
在分析级方法中淘汰HFIP
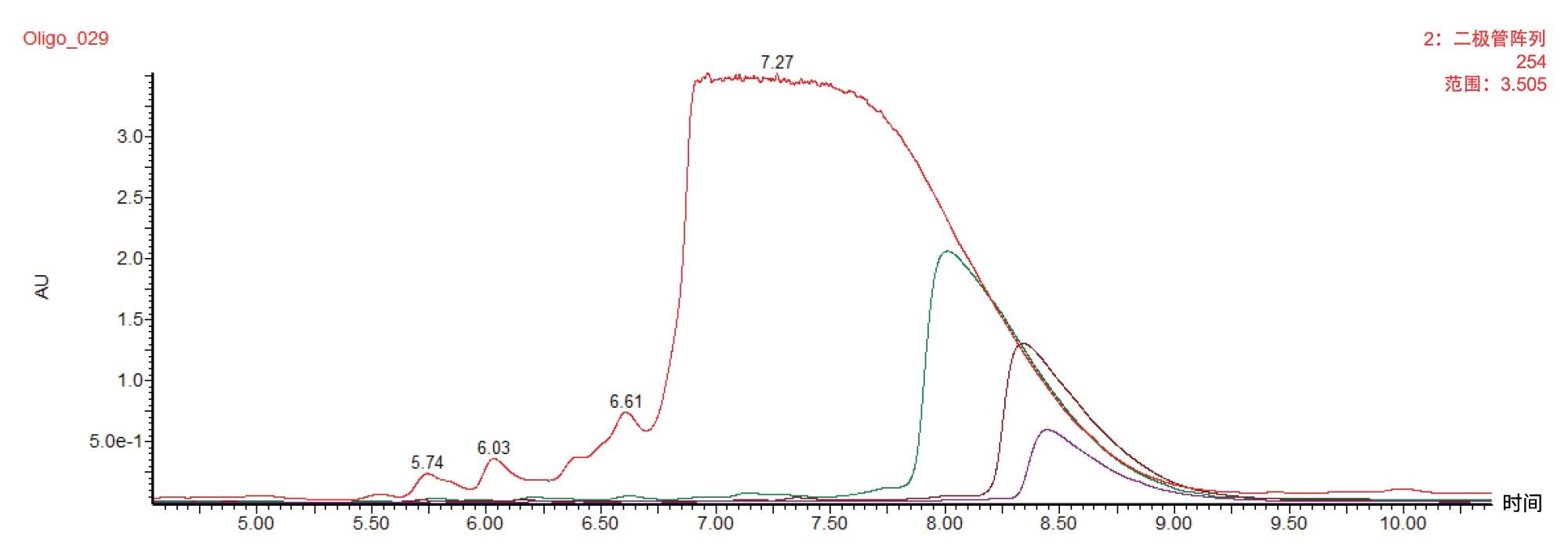
首先使用ACQUITY UPLC BEH C18 1.7 µm 2.1 × 100 mm寡核苷酸分析专用柱对20 mer寡核苷酸进行分析级分离。该方法采用HFIP作为流动相,柱温为60 °C。图1展示了在这些条件下获得的示例色谱图。采用60 °C的柱温是为了消除二级结构的影响并提高寡核苷酸的扩散性。由于大多数短链寡核苷酸缺乏二级结构,它们在室温下即可被充分分离,从而大大简化了向制备级规模的放大过程。
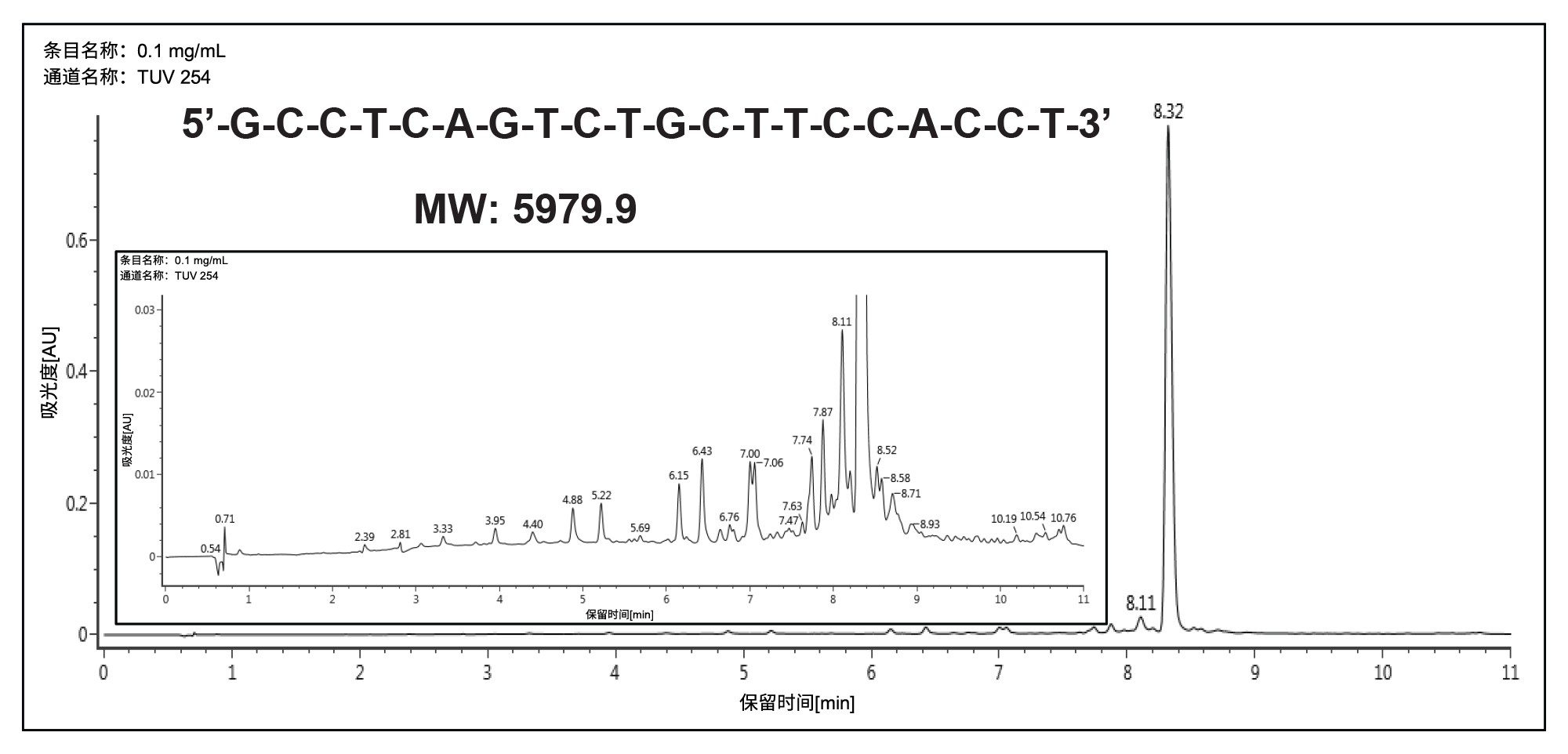
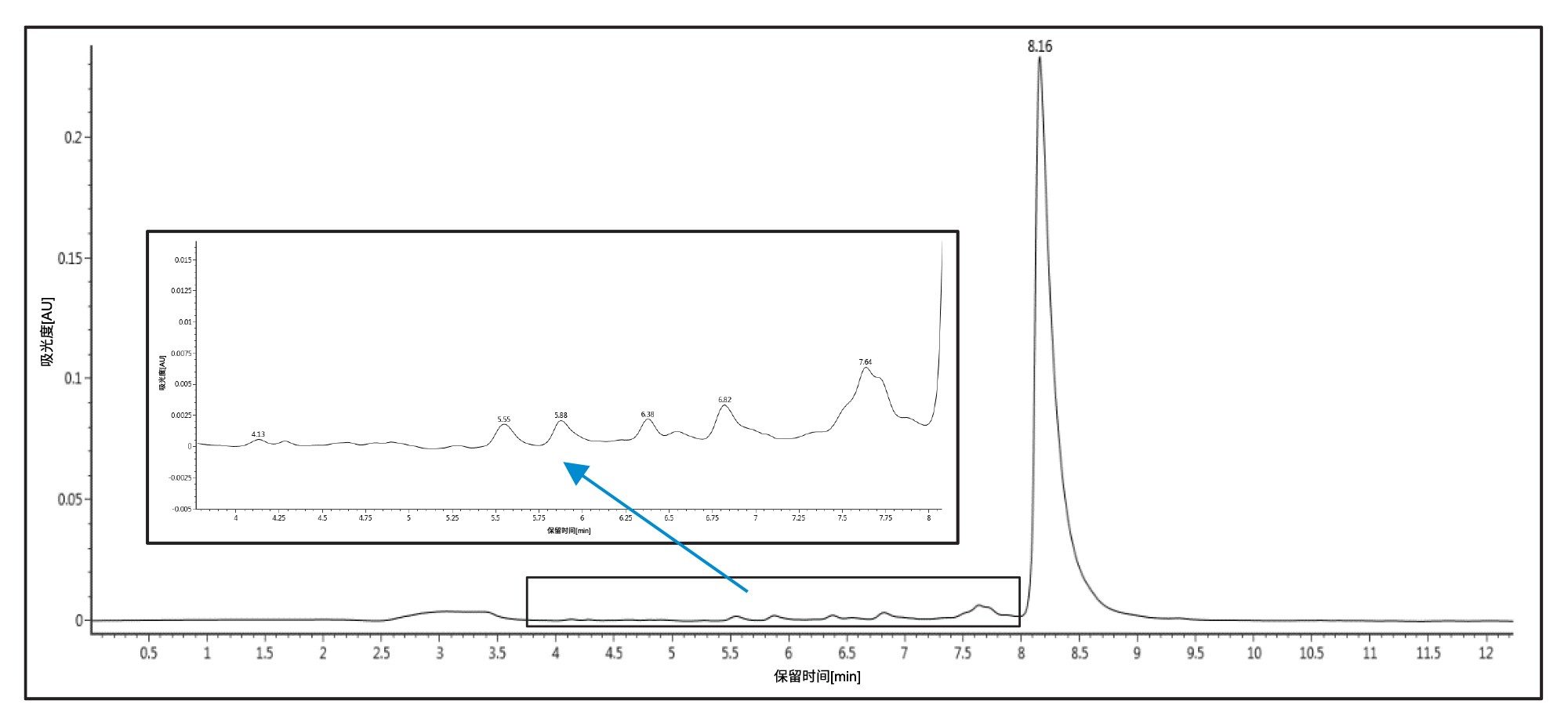
在下一轮实验中,我们使用三乙基乙酸铵(TEAA)开发出一种分析级方法。该方法使用4.6 × 50 mm XBridge™ BEH C18 2.5 µm寡核苷酸分析专用柱,流动相A为100 mM TEAA (pH 7.0),流动相B为乙腈。柱温保持在25 ℃,寡核苷酸以多步梯度洗脱,流动相B起始比例为5%。然后在0.25 min内,流动相B从5%增加至8%,接着在9.75 min内从8% B增加到12% B,最后在0.1 min内从12% B增加到95% B。保持95%的流动相B 0.9 min,然后在0.1 min内恢复到5% B。在下一次进样之前,色谱柱在5%流动相B下平衡4.9 min。图2展示了获得的色谱图。尽管使用TEAA的n-1和n-x分离效率不如初始运行TEA/HFIP时的效率,但这种分离仍然能够将目标峰与终止失败序列分离。该方法提供了一种经济有效、危害性较低的溶剂体系,同时保持了溶剂挥发性,便于进行后续的馏分收集和干燥步骤。
缩放至制备型大孔径色谱柱
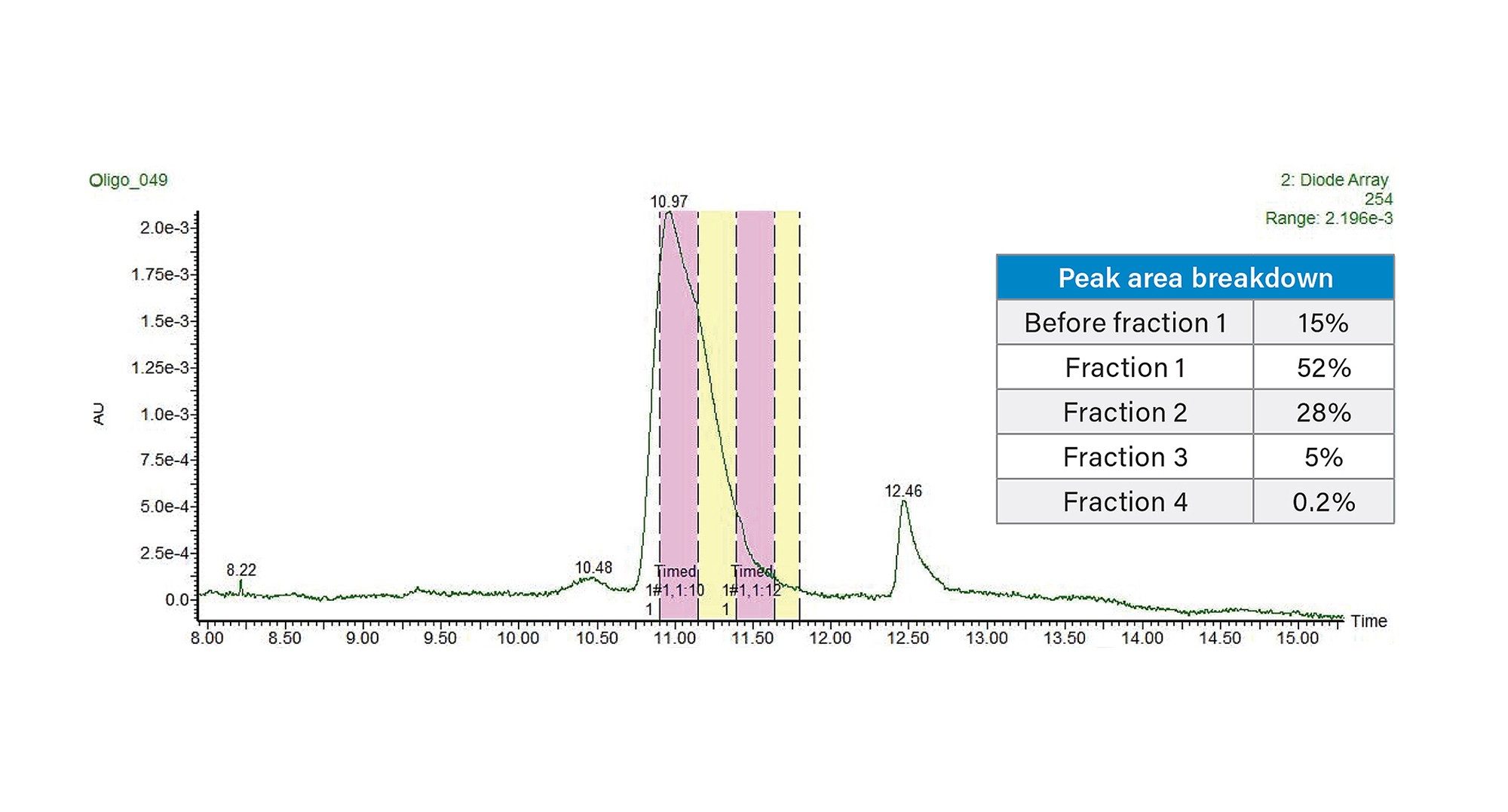
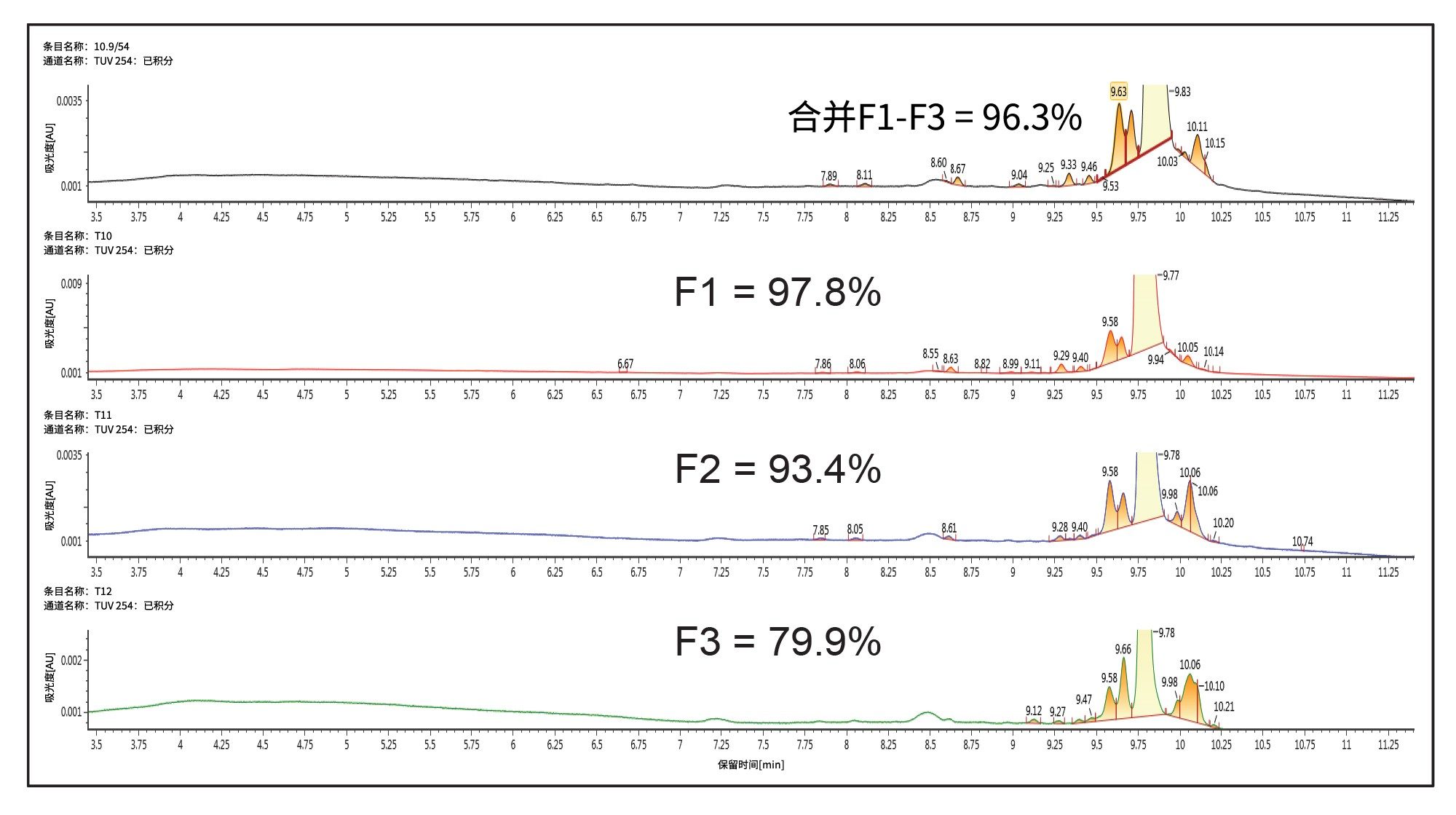
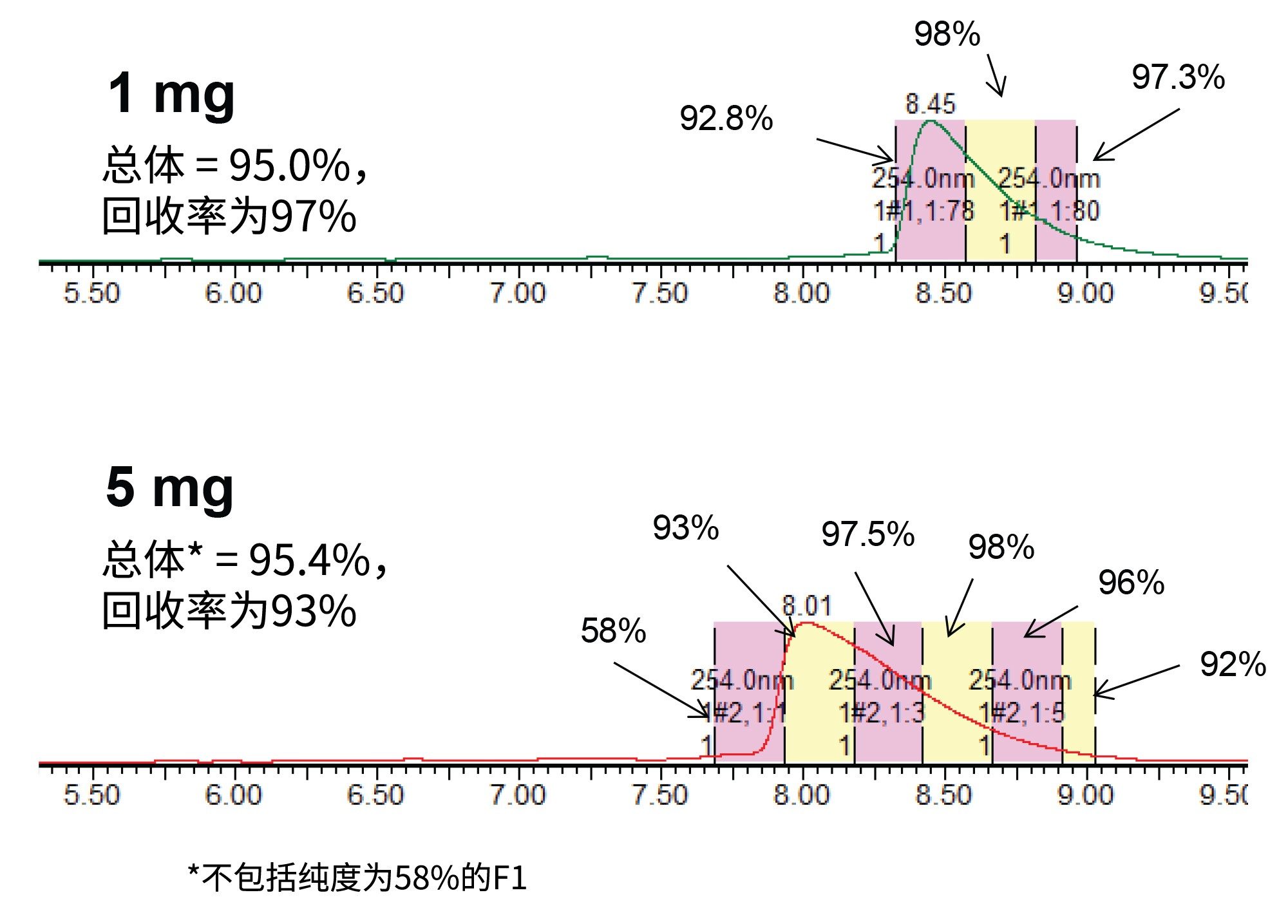
然后,使用Waters™梯度计算器和Waters AutoPurification系统(包括3767自动进样器/馏分收集器、SFO、UV检测器(PDA或TUV)和SQD2单四极杆MS,以及用于MS的ACQUITY ISM补充流泵)将这种使用TEAA的新分析方法转移至30 × 50 mm XBridge BEH C18 OBD 2.5 µm寡核苷酸专用制备柱上。这部分工作的目的是评估峰的纯度,因此采用基于时间的收集模式。但是,也可以使用基于MS的馏分收集方法,该方法通常通过仅收集目标质量数来提高选择性。在不同色谱柱内径之间缩放方法时,需要考虑的最重要的参数之一是色谱柱流速,它与色谱柱半径的平方成正比。此外,将方法从4.6 mm内径(ID)色谱柱放大至30 mm ID色谱柱时,基于色谱柱体积比,放大后的载样因子为42倍。因此,10 µg分析级进样量可转换为410 µg制备级进样量。在该载样量下获得的制备级色谱图如图3所示。通过设置时间触发馏分收集,每管收集洗脱液15 s。使用初始UPLC方法对收集到的单个馏分和混合馏分进行分析,结果表明,3种主要馏分中,单个馏分的纯度范围为98%~80%(在图3中标记为F1~F3),合并纯度为96.3%。图4展示了这些馏分的分析级纯度分析结果。计算出的混合馏分总回收率为85%。请注意,如果需要提高纯度,建议不要合并第三个馏分。F1 + F2的合并馏分纯度超过97%,而回收率将受到约5%的影响。
增加载样量
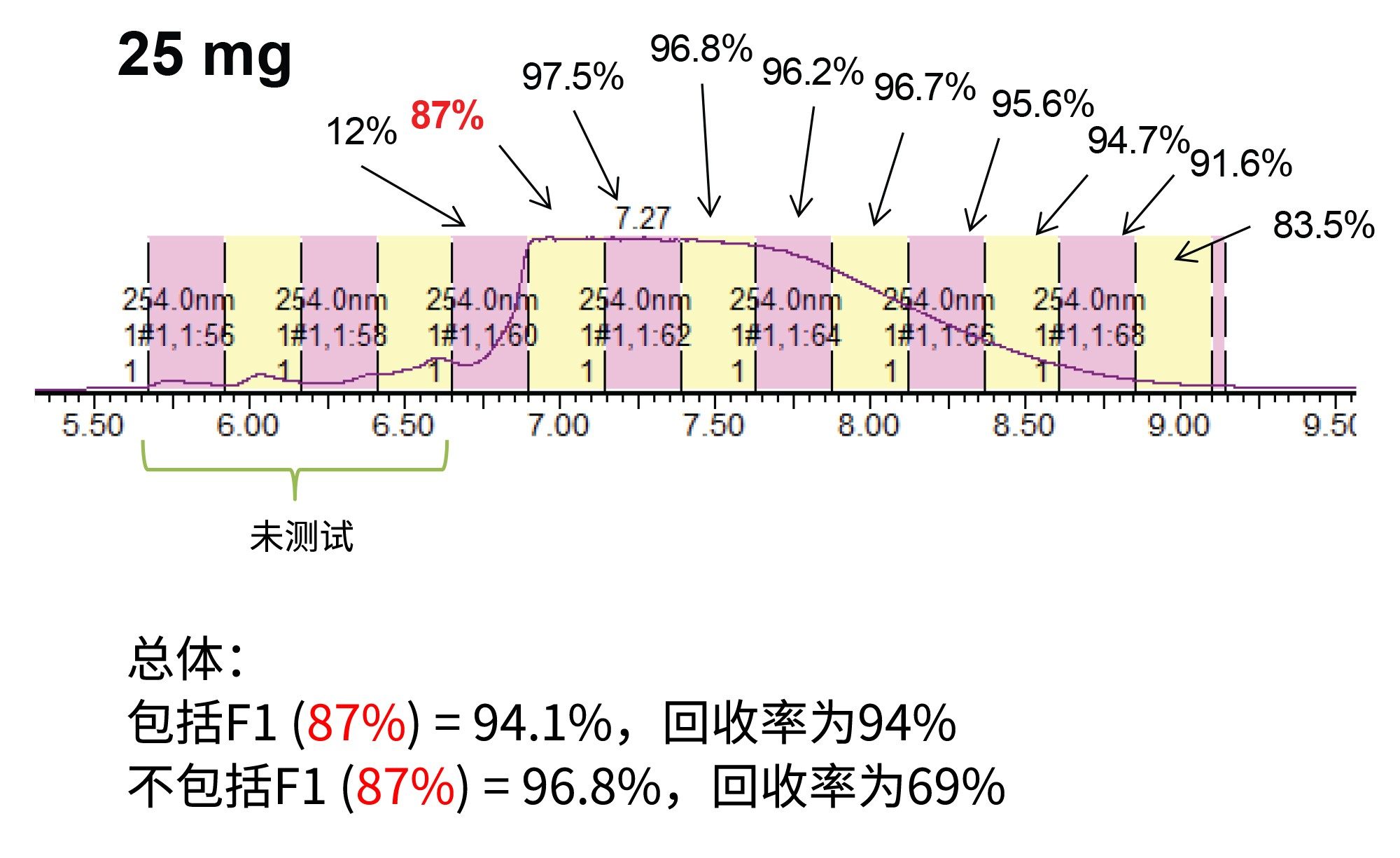
图3和图4展示的纯化结果表明,稳健的分析级分离方法可放大至30 mm制备级色谱柱,且性能不受影响。然而,衡量纯化方法成功与否的另一个标准是生产率。该方法每小时可产生约1 mg的纯化寡核苷酸。提高生产率的方法之一是增加每次进样的载样量。该方法的挑战在于,由于会出现与上样量相关的峰展宽,可能会在获得纯化物质时损失分离度或回收率。图5展示了载样量增加到(25 mg)的50倍时,30 × 50 mm 2.5 µm寡核苷酸RPLC色谱柱上的色谱分离所受到的影响。与预期一样,随着上样量的增加,峰起点进一步前移。
评估峰纯度
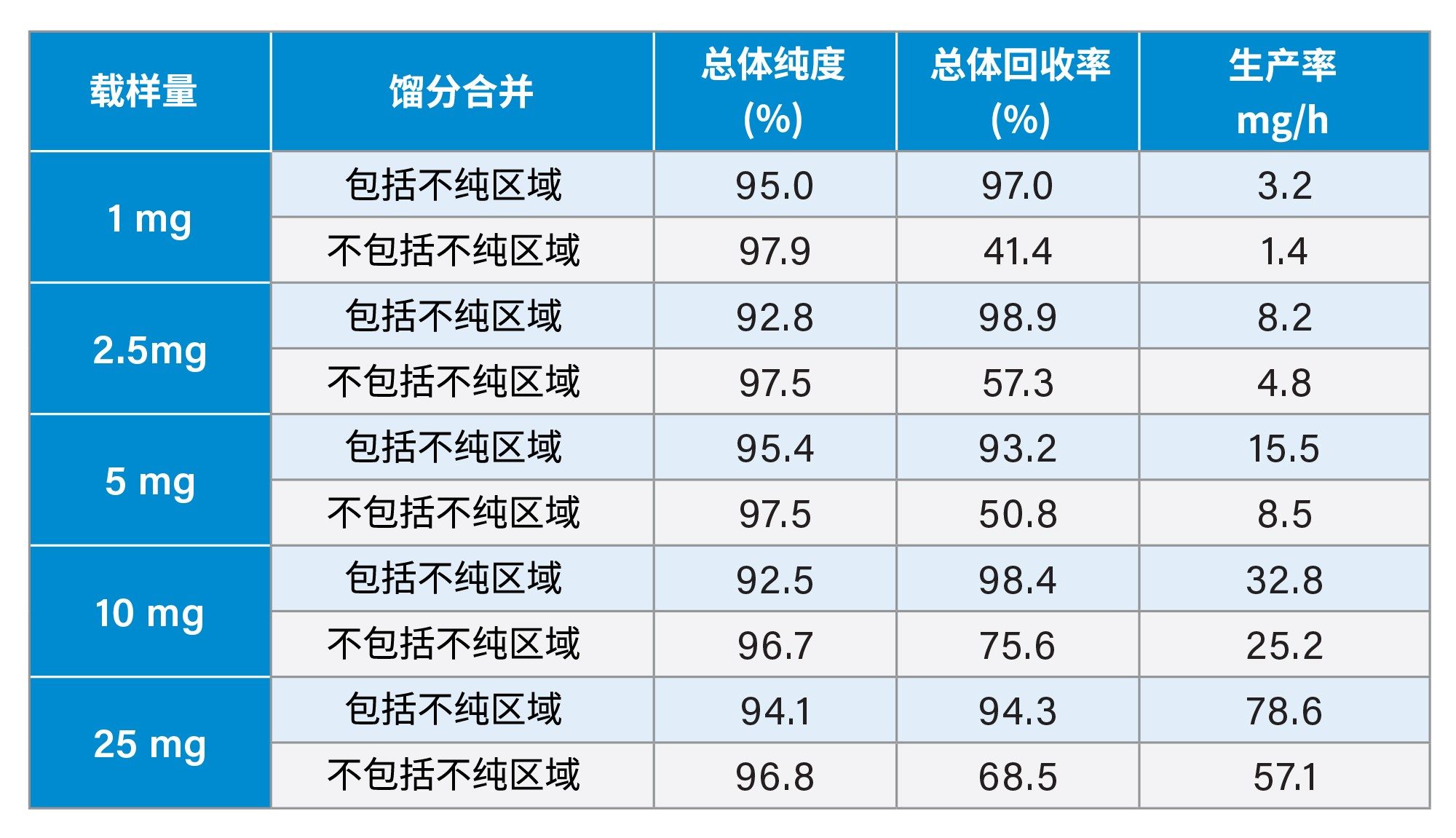
如图5所示,每次进样以15 s为时间间隔收集馏分,浓缩后通过UPLC HFIP/TEA方法进行分析。这些纯度测定结果展示在图6和图7中。正如之前报道的那样,从峰起点到峰顶点的区域纯度明显低于峰的其他区域3。 然而,随着载样量的增加,峰展宽,纯馏分的数量也随之增加,从而提高回收率和生产率。根据总体纯度要求,可以将必要的试管合并以得到最终的纯化物质。表1显示了不同载样量和不同分馏方法所实现的生产率的计算结果。
方法参数
为了进一步了解影响生产率的变量,研究中还对其他纯化条件进行了考察。较低的流速可以改善分离效果,但会降低生产率。研究中评估了梯度流速从17 mL/min增加到25 mL/min的影响。表2显示纯度略有下降,但生产率提高了50%。
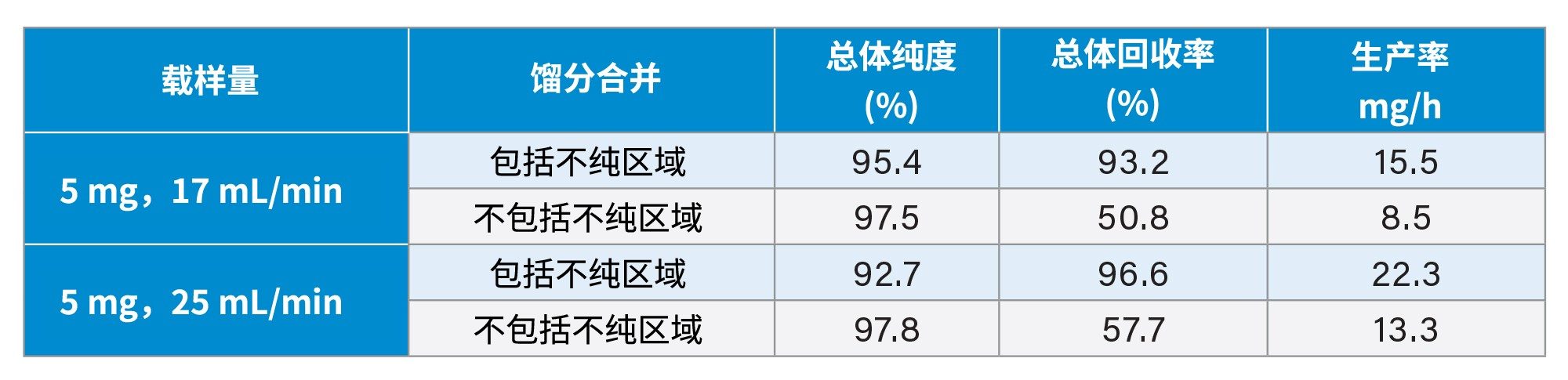
另一种优化生产率的方法是应用聚焦梯度。前面的示例已经应用了聚焦梯度,因此我们研究了在洗脱窗口之外增加流速的情况。在再生和再平衡步骤中提高流动相流速可节省大量时间。
此外,方法运行时间也略有缩短,利用自动进样器清洗和进样下一个样品的时间来平衡色谱柱。在本例中,运行时间减少了1分钟。结合流速调整,整体生产率提高了约25%4。 优化后的制备型色谱方法如表2所示。该方法可根据所需纯度,以75~100 mg/h的速度生成纯化寡核苷酸。
最后,使用其他色谱柱尺寸,特别是孔径更宽的色谱柱,这一点值得考虑。这样可以进一步提高生产率,但本研究未对此进行测试。将色谱柱内径从30 mm增加到50 mm可使载样量和生产率提高大约3倍,但需要使用47.2 mL/min的速率。另一个提高生产率的方法是实施离线色谱柱清洗和平衡4。 此方法需要额外的硬件,包括切换阀、再生泵和第二根色谱柱。但是,这完全省去了方法中的这段时间。
结论
本应用纪要介绍了高效分析纯度测量方法与高生产率纯化方法之间的相互作用和可扩展性。虽然HFIP流动相适用于分析级LC-MS纯度测定,但在纯化过程中却存在问题。我们展示了如何将采用HFIP流动相的分析级分离成功转换为在大孔径制备柱上进行基于TEAA的纯化运行。通过优化方法参数和载样量,我们设计出了一套简单的实验室规模纯化方案,能够以超过100 mg/h的速度产出纯度超过95%的纯化物质。通过增加制备色谱柱的内径以及实施离线色谱柱清洗和再生,还可实现进一步的速度提升。我们希望这些技术对该行业有价值,因为它们会采取新的举措来支持不断扩张的合成寡核苷酸行业。
参考资料
- Patrik D. McDonald et al., 最佳柱床密度[OBD™]色谱柱:实验室级分离和纯化的关键技术, 720001939ZH.
- Gilar, M. et al., 离子对反相液相色谱法分析寡核苷酸的最佳实践:色谱柱及其填料, 720006948ZH.
- Gilar, M. et al., HPLC and UPLC column for Analysis of Oligonucleotides, Waters 720002376.
- Lefebvre, P. et al., Evaluating the Tools for Improving Purification Productivity.Waters application note 720001696.2007
720008266ZH,2024年2月