通过LC-MS/MS分析食品中28种EU监管和推荐检测的PFAS – 第1部分:蔬菜、水果和婴儿食品
摘要
本研究介绍了一种针对蔬菜、水果和婴儿食品中的全氟烷基和多氟烷基化合物(PFAS)进行全面分析的优化LC-MS/MS方法。该方法的定量限非常低,对于基质中的某些化合物可低至0.0005 µg/kg,同时能够准确检测和定量欧盟委员会(EU) 2022/1431号法规建议中列出的PFAS化合物。该方法结合使用了灵敏度增强的Xevo™ TQ Absolute MS系统与净化效力更高的GCB/WAX双层双相原型SPE小柱,表现出优异的回收率,对于强制检测的PFAS可达到87%~116%,而对于大多数推荐检测的化合物可达到65%~131%,且重复性(RSDr)≤10%。
优势
- 使用高灵敏度定量方法分析欧盟委员会建议中要求强制检测、推荐检测和考虑检测的PFAS,可将所有PFAS纳入符合要求的单一方法中
- 在蔬菜、水果和婴儿食品中的定量限极低(低至0.0005 µg/kg),优于EURL POPs指南中规定的标准
- 优异的方法回收率和重复性,符合指南中的可接受标准
- 使用含有GCB和WAX的新型双层双相SPE小柱净化样品,使FOSA获得良好的回收率,从而有效净化中性PFAS
- 与之前的UPLC™方法相比,时间效率更高,样品分析时间缩短50%,同时确保直链和支链PFAS化合物与潜在干扰物之间实现基线分离
简介
全氟烷基和多氟烷基化合物(PFAS)是一类合成化合物,广泛用于各种工业和商业用途,以其持久性和潜在健康危害而闻名。由于PFAS存在于农业环境和食物链中,对消费者的健康构成潜在风险,因此,监管机构越来越重视严格监测和控制食品中的PFAS含量。
在欧盟(EU),欧盟委员会(EU)第2022/2388号法规(修订了(EC)第1881/2006号法规,自2023年1月1日起实施)规定了动物源性食品中PFOS、PFOA、PFNA和PFHxS各化合物的最高限量,以及这些PFAS总和的最高限量1。 而2022年9月生效的欧盟委员会(EU) 2022/1431号法规建议则建议各成员国在2022、2023、2024和2025年间检测种类更广泛的食品中相同的四种PFAS,而不仅限于第2022/2388号法规中规定的动物源性食品。此外,还建议在各种食品中监测更多种类的PFAS2。
2022年9月生效的欧盟委员会(EU) 2022/1428号实施条例(Commission Implementing Regulation (EU) 2022/1428, CIR)规定了为控制某些食品中PFAS的采样和分析方法,还规定了方法验证的可接受标准,以及关于结果报告和解释的信息3。 此外,EURL POPs于2022年发布了针对PFAS方法的指导文件,提供了有关预期方法性能和定量限的信息4,5。 AOAC标准方法性能要求(Standard Method Performance Requirements, SMPRs®)描述了在方法评估过程中建议使用的最低性能特征。评估方式可以是现场确证、单实验室验证或多地点合作研究。AOAC SMPRs可用作欧盟委员会(EU) 2022/1428号实施条例规定以外情况下确证的可接受标准。AOAC SMPR 2023.003已发布,其中规定了针对各种食品中PFAS分析的方法性能要求6。
本研究的第1部分重点关注如何针对蔬菜、水果和婴儿食品中超痕量水平的PFAS开发一种灵敏的LC-MS/MS检测和定量方法,满足对可靠分析方法的需求,并符合近期指南中规定的标准。第2部分重点关注动物产品中PFAS的测定。
实验
标准品和溶液
所有标准品均购自Wellington Laboratories。使用以下标准品制备储备液:

使用甲醇制备天然PFAS混标储备液(每种分析物各500 ng/mL),然后进行连续稀释。用甲醇制备提取内标(EIS)混标溶液(每种标记分析物各1 ng/mL),用于在提取前加标食品样品。使用1:1水:甲醇(本研究中的所有比例均为v:v)制备EIS和进样内标(IIS)的混标。EIS + IIS混标中每种标记分析物各0.2 ng/mL;该溶液用作标准曲线的稀释剂。最后,用1:1水:甲醇制备IIS混标溶液(每种标记分析物各20 ng/mL),用于在提取和净化后加标各LC样品瓶。
制备0.00125–5 ng/mL(相当于实际样品中0.00025-1 µg/kg/g)范围内的溶剂标准曲线,用于样品分析。
样品前处理
在实验室进行任何操作之前,必须遵循良好操作规范,防止或尽可能减少环境和试剂中的PFAS污染(详细信息请参见白皮书720007905)7。
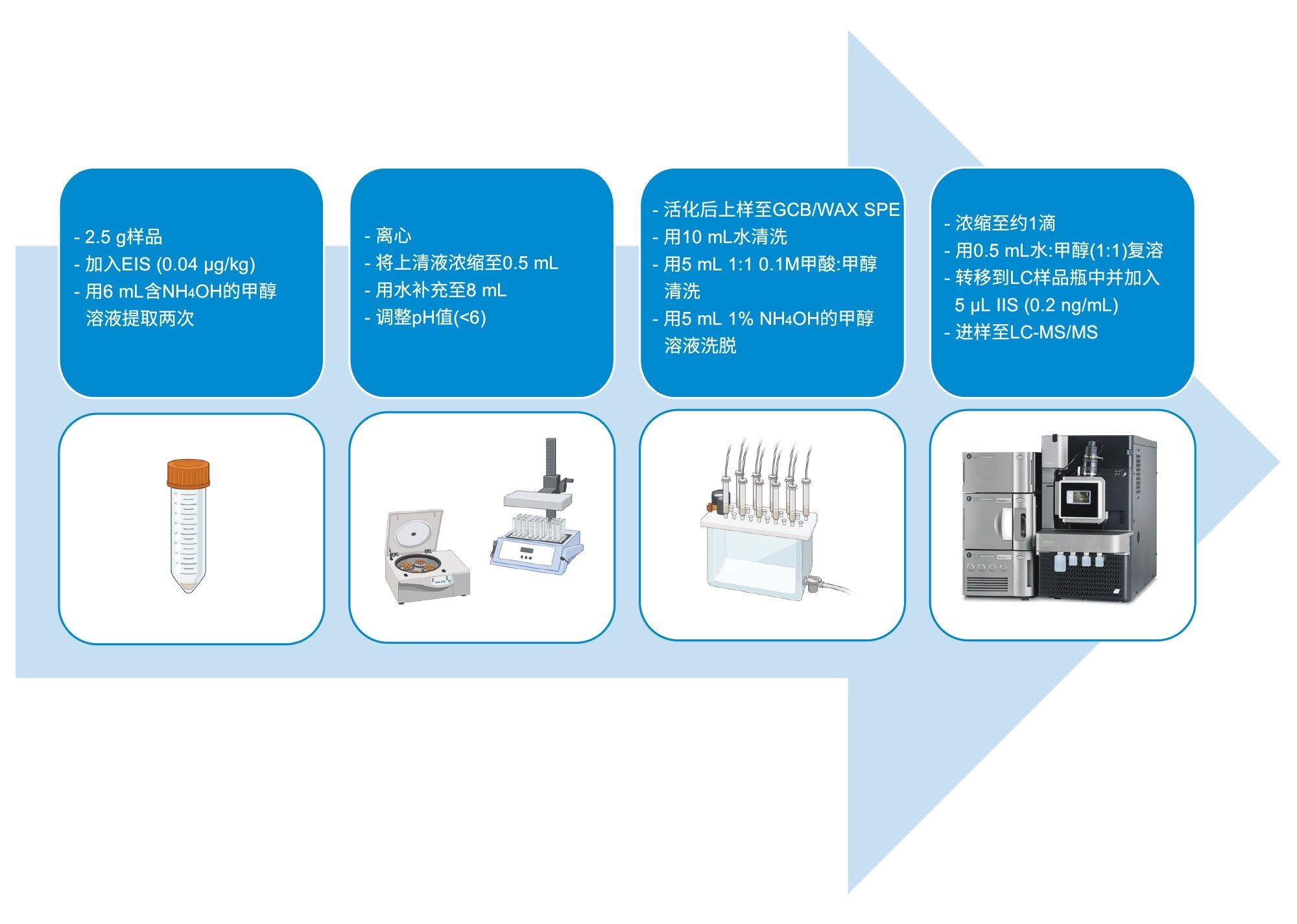
供试品包括蔬菜(番茄)、水果(苹果)和婴儿食品(由水果和酸奶制成的有机柔滑果泥),均购自当地杂货店。将番茄和苹果的可食用部分切成薄片,并使用厨房搅拌机均质化处理。样品储存于冷冻柜(-20 °C)中,临提取前置于冰箱(4 °C)中过夜解冻。
取2.5 g样品置于15 mL Falcon离心管中,加入100 μL EIS(浓度为0.04 µg/kg),涡旋几秒钟。
用6 mL 0.3%氢氧化铵甲醇溶液提取样品,将试管涡旋混合10分钟。以5000 g离心后,将上清液定量转移至干净的空管中。重复提取步骤,合并约12 mL提取液,在50 ºC的温和氮气流下浓缩至0.5 mL。使用试剂用水将各样品复溶至8 mL。使用试纸检查各样品的pH值,并通过添加几滴50%甲酸溶液(如果需要)进行校正,确保复溶提取液的pH值低于6个单位。
将提取液上样至含有弱阴离子交换(WAX)和石墨化炭黑(GCB)吸附剂的沃特世双层双相原型SPE小柱中。这款小柱的GCB层位于WAX层上方,用于净化食品样品。提前用15 mL 1%氢氧化铵甲醇溶液和5 mL 0.3 M甲酸水溶液活化小柱。上样后,用10 mL试剂用水和5 mL 1:1 0.1 M甲酸:甲醇清洗小柱。将小柱干燥10秒,然后用5 mL 1%氢氧化铵甲醇溶液洗脱分析物,用干净的Falcon管收集洗脱液。
在50 ºC的温和氮气流下将洗脱液浓缩至约1滴,然后用0.5 mL 1:1水:甲醇复溶。复溶分两步进行:先向管中加入250 μL甲醇,涡旋混合;然后向管中加入250 μL水,再次涡旋混合。此方法旨在提高弱极性PFAS的溶解度。
将净化后的复溶样品(0.5 mL)转移至带有聚乙烯盖(P/N:186000305)的聚丙烯LC样品瓶(P/N:186005219)中。向每个样品瓶中加入5 μL IIS溶液(样品瓶中每种标记IIS各0.2 ng/mL),然后涡旋混合,并放入自动进样器中以备进样。方法示意图见图1,得到的浓度因子为5。
液相色谱条件
|
液相色谱系统: |
ACQUITY™ PREMIER UPLC,配备PFAS分析方法包 |
|
样品瓶: |
聚丙烯自动进样器样品瓶(P/N:186005219),带有聚乙烯盖(P/N:186000305) |
|
分析柱: |
ACQUITY Premier UPLC BEH™ C18, 2.1 × 50 mm, 1.7 µm(P/N:186009452) |
|
隔离柱: |
Atlantis™ Premier BEH C18 AX 2.1 × 50 mm, 5.0 µm(P/N:186009407) |
|
柱温: |
35 °C |
|
样品温度: |
10 °C |
|
进样体积: |
5 µL |
|
流速: |
0.3 mL/min |
|
流动相A: |
2 mM乙酸铵水溶液 |
|
流动相B: |
含2 mM乙酸铵的甲醇/乙腈1/1 (v/v) |
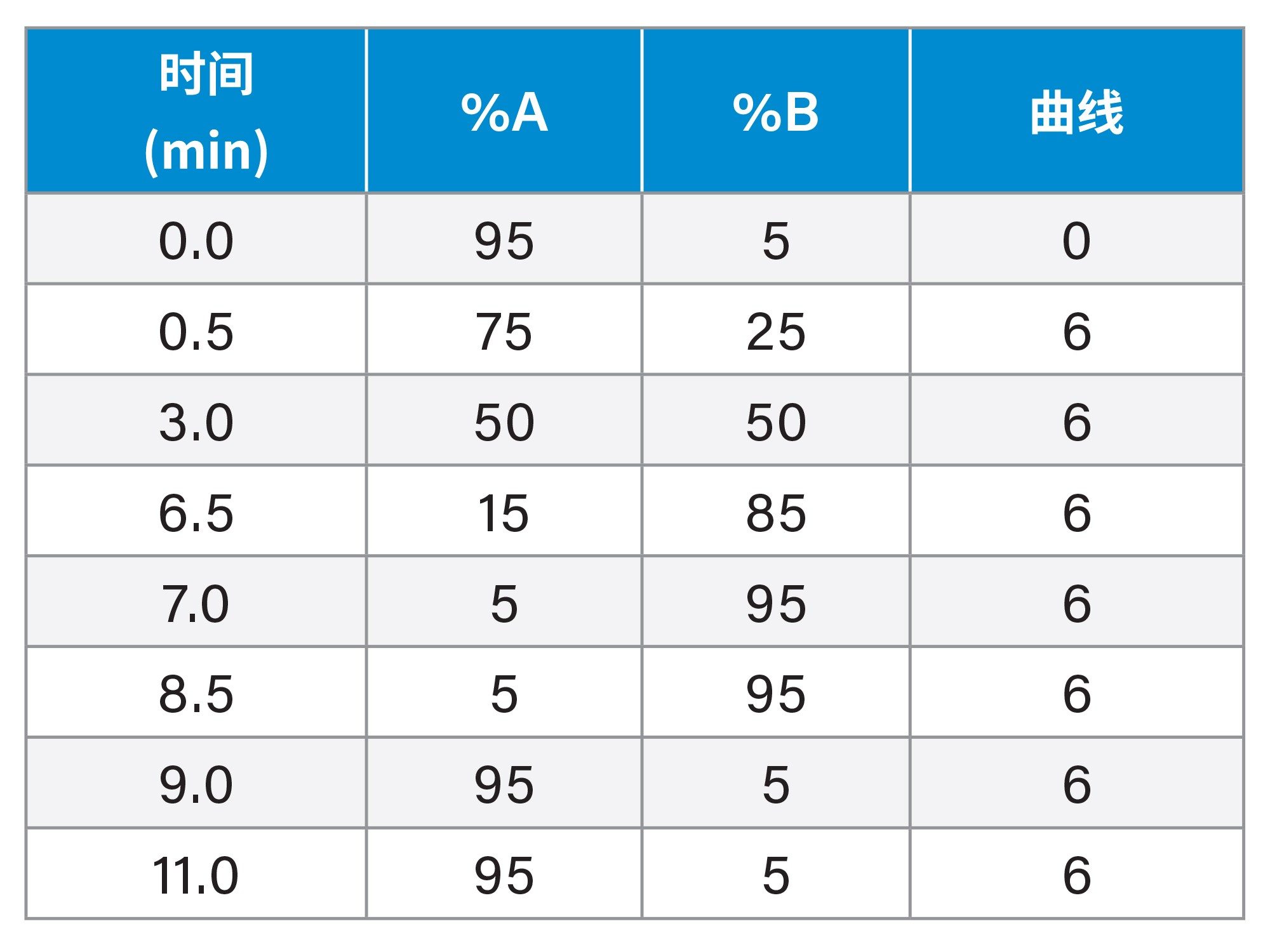
梯度表
MS条件
|
质谱系统: |
Xevo TQ Absolute |
|
电离模式: |
电喷雾负离子模式 |
|
离子源温度: |
100 °C |
|
毛细管电压: |
0.5 kV |
|
脱溶剂气温度: |
350 °C |
|
脱溶剂气流速: |
900 L/h |
|
锥孔气体流速: |
150 L/h |
|
MRM方法: |
有关完整MRM方法的详细信息,请参见附录 |
数据管理
|
软件: |
waters_connect™定量软件平台 |
方法性能评估
使用EURL POPs PFAS指导文件和AOAC SMPR中规定的标准评估方法性能4,6。 对于批内方法性能,通过向番茄、苹果和婴儿食品样品中加入28种天然PFAS的混合物,三个浓度水平一式三份,评估正确度。然后按照上一节所述的方法提取并分析溶剂空白、程序空白、基质空白和加标样品。根据批内重复性条件下的相对标准偏差评估方法重复性(RSDr)。计算各浓度水平的表观回收率和RSDr:
水平1:对应于方法定量分析下限(m-LLOQ)
水平2:对应于10倍m-LLOQ
水平3:对应于方法定量分析上限(m-ULOQ)
每个批次的分析序列均由两组不同的溶剂校准物(每条曲线≥6个点,不包括空白样品)组成,这些校准物将实测样品和加标样品夹在中间。
结果与讨论
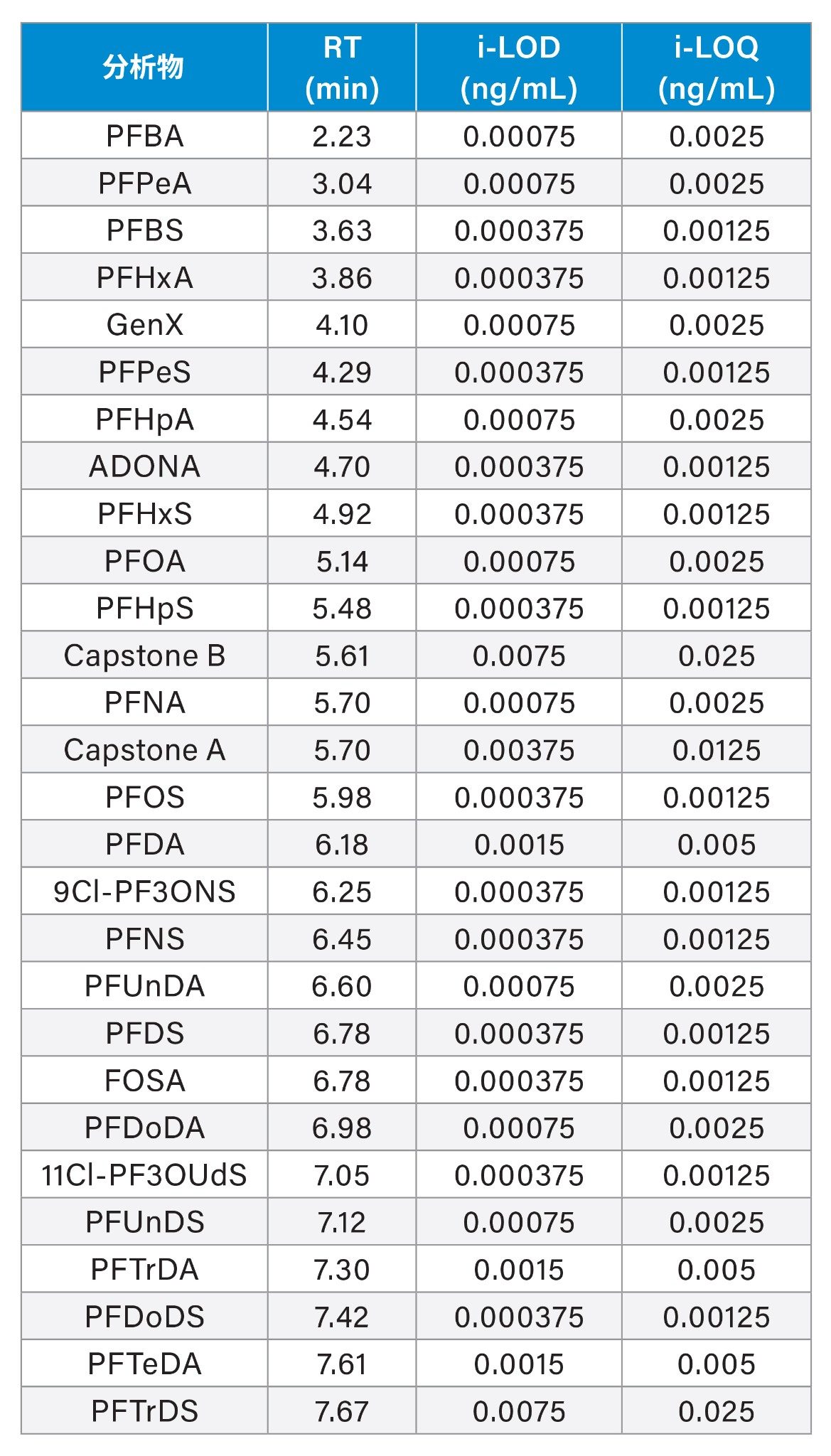
该方法涵盖了欧盟委员会(EU) 2022/1431号法规建议中列出的28种PFAS,包括强制检测、推荐检测和“考虑检测”的PFAS(PFAS列表见表1)。
在样品提取之前加入EIS,用于校正天然化合物的提取回收率和基质效应。在样品净化后加入IIS,用于校正提取标准品的复溶变化、基质效应和潜在进样变化。有了EIS和IIS,常规样品分析就无需进行基质匹配。这种方法已在之前的研究中讨论过8。
色谱分析
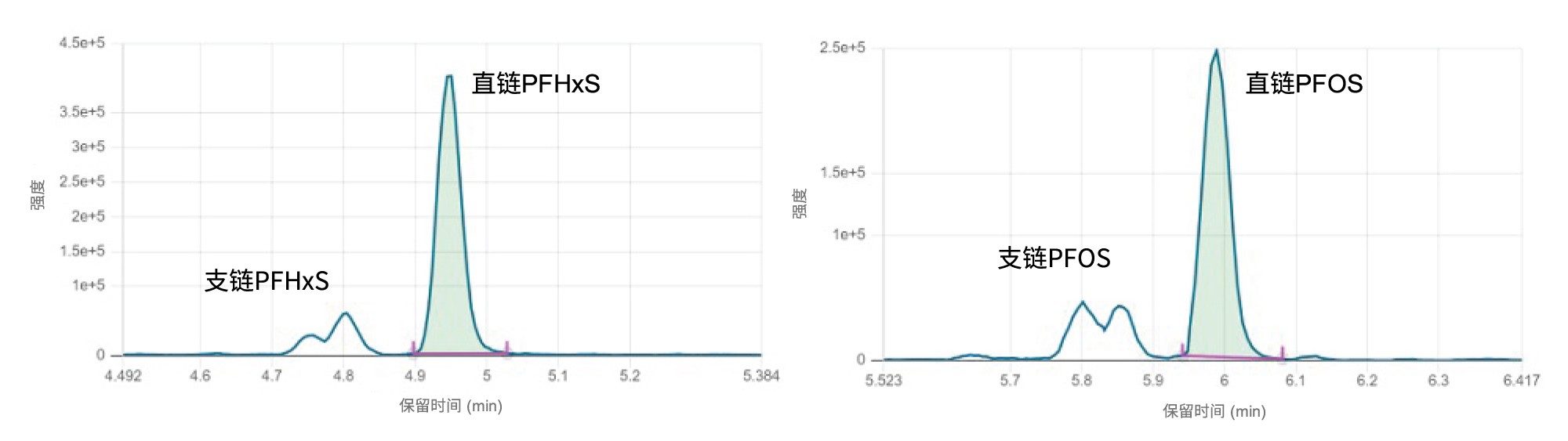
将之前开发的LC方法缩放后,运行时间从22 min缩短至11 min,使用50 mm BEH C18色谱柱实现了直链和支链PFAS的基线分离。利用甲醇和乙腈的有机流动相组合,使PFAS与某些食品类型中常见的潜在干扰物实现了基线分离(例如胆酸与PFOS的分离)。图2显示了PFHxS和PFOS的色谱图,从图中可以看出,由于实现了出色的色谱分离,直链和支链PFAS既可以单独定量,也可以测定总含量。有关LC优化的更多信息可参阅之前的研究应用纪要720008108ZH9。
分析物的色谱保留时间(RT)与IS的RT之比(即分析物的相对RT)与校正标准品的RT之比对应,偏差≤1%。
第一个洗脱的化合物(PFBA,RT=2.23 min)的RT大于色谱柱死体积时间(t0=0.37)的6倍,因此,本研究中分析的所有PFAS均获得了足够的保留。
线性和定量限
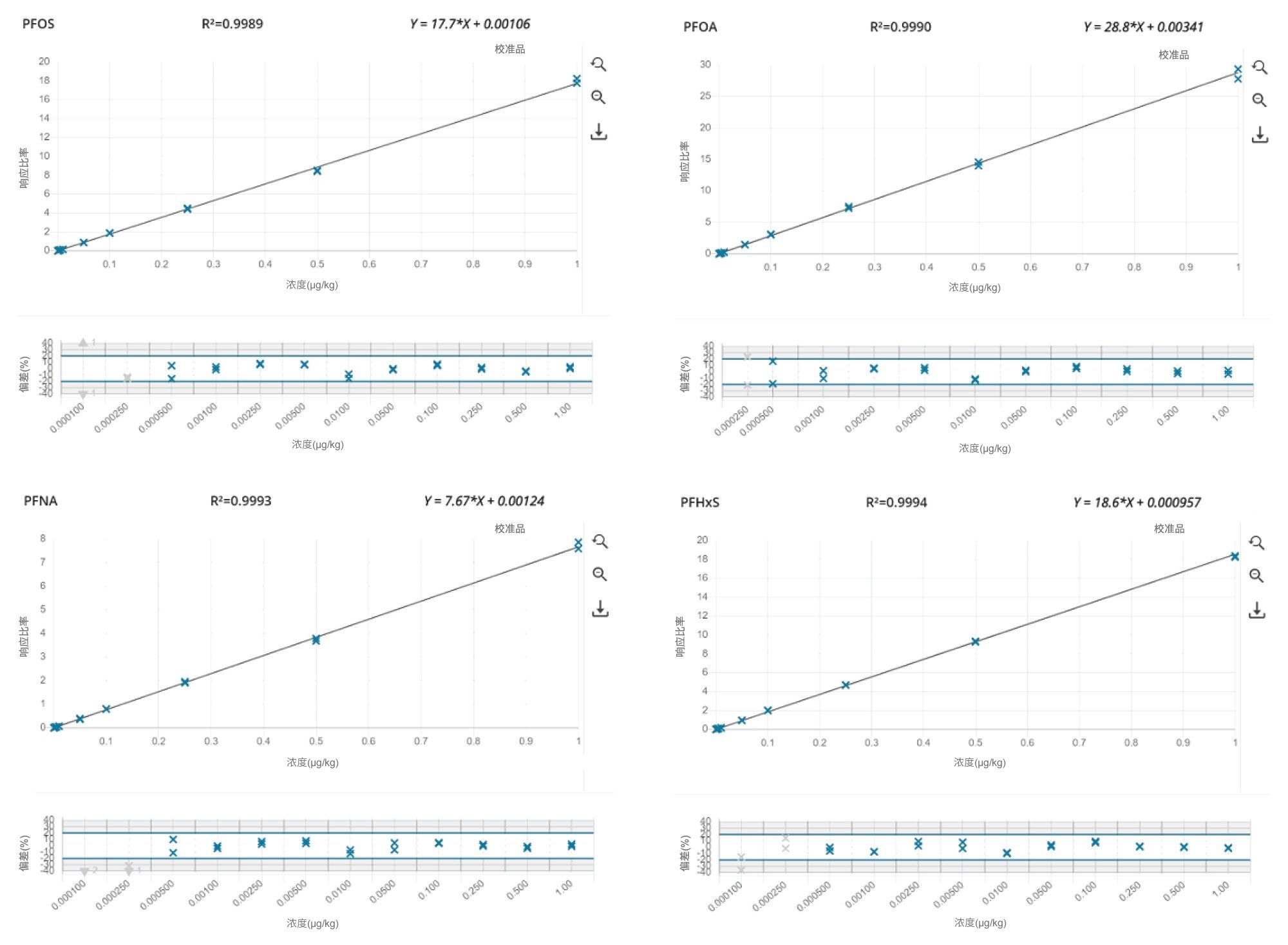
使用1/x加权因子绘制溶剂标准品的响应(将实测样品和加标样品夹在中间),构建标准曲线。不同分析物和基质的方法线性范围不同。在大多数情况下,标准曲线的决定系数(R2)均高于0.99,残差均低于±20%。样品提取物的离子丰度比在同一序列中所有校准标准品离子丰度比平均值的±30%(相对)范围内。
仪器检测限(i-LOD)10根据溶剂标准品计算得出(在LOD水平下,信噪比≥3),某些单个PFAS的i-LOD低至0.0004 ng/mL(相当于柱上进样量2 fg)。
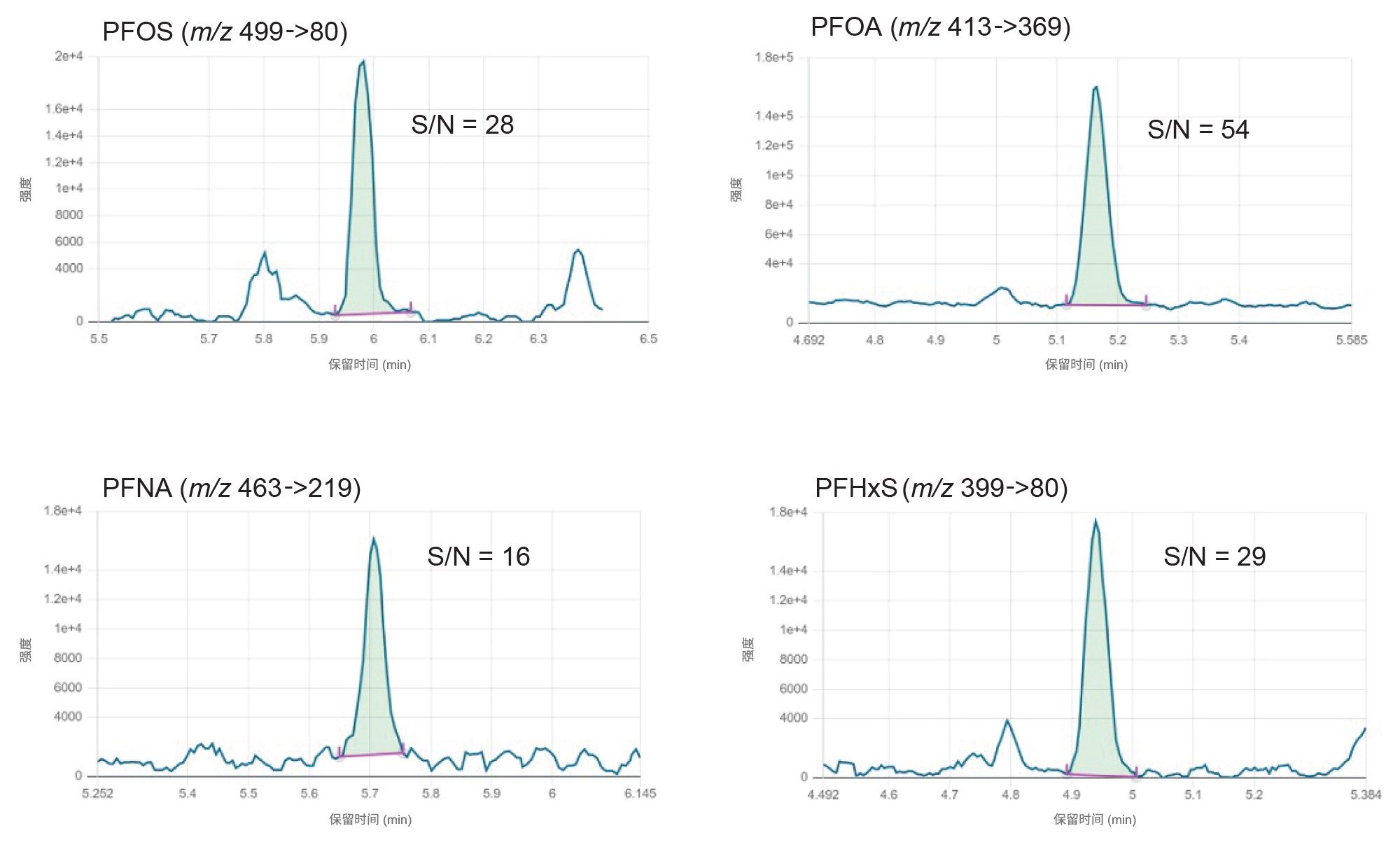
方法定量下限和方法定量上限(m-LLOQ和m-ULOQ)分别作为校准范围的最低和最高浓度,前提是m-LLOQ下的信噪比≥10(噪音处理=峰到峰)。对供试品的m-LLOQ低至0.0005 µg/kg。i-LOD、i-LOQ和保留时间见表1,对各食品商品的m-LLOQ、方法线性范围和线性见表2、3和4。
值得注意的是,在检测的所有食品商品中,该方法能够对低至0.0005 µg/kg的四种强制检测PFAS(PFOS、PFOA、PFNA和PFHxS)进行准确定量。图3显示了标准曲线和残差图,图4显示了m-LLOQ水平的示例色谱图。
正确度和重复性
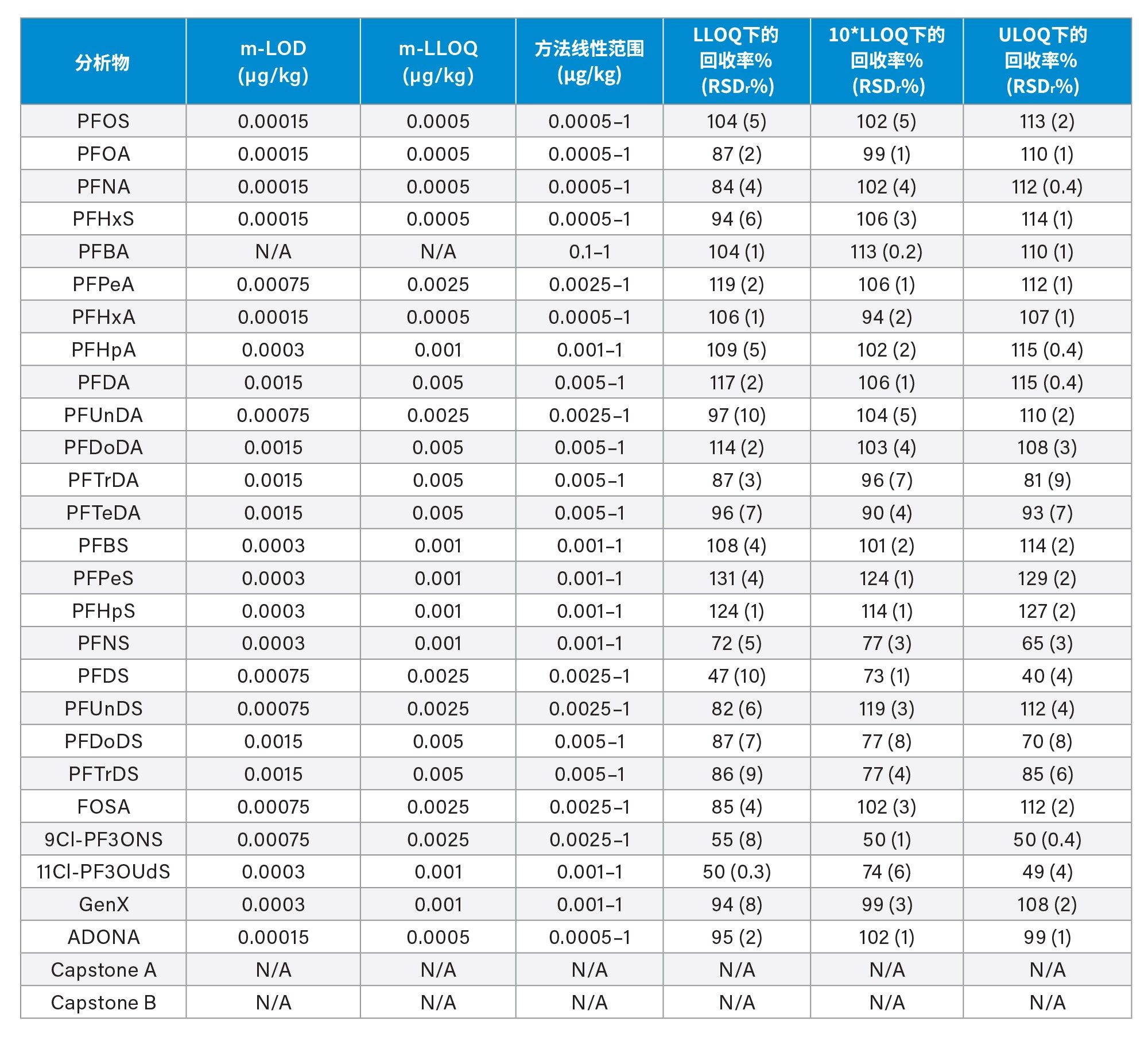
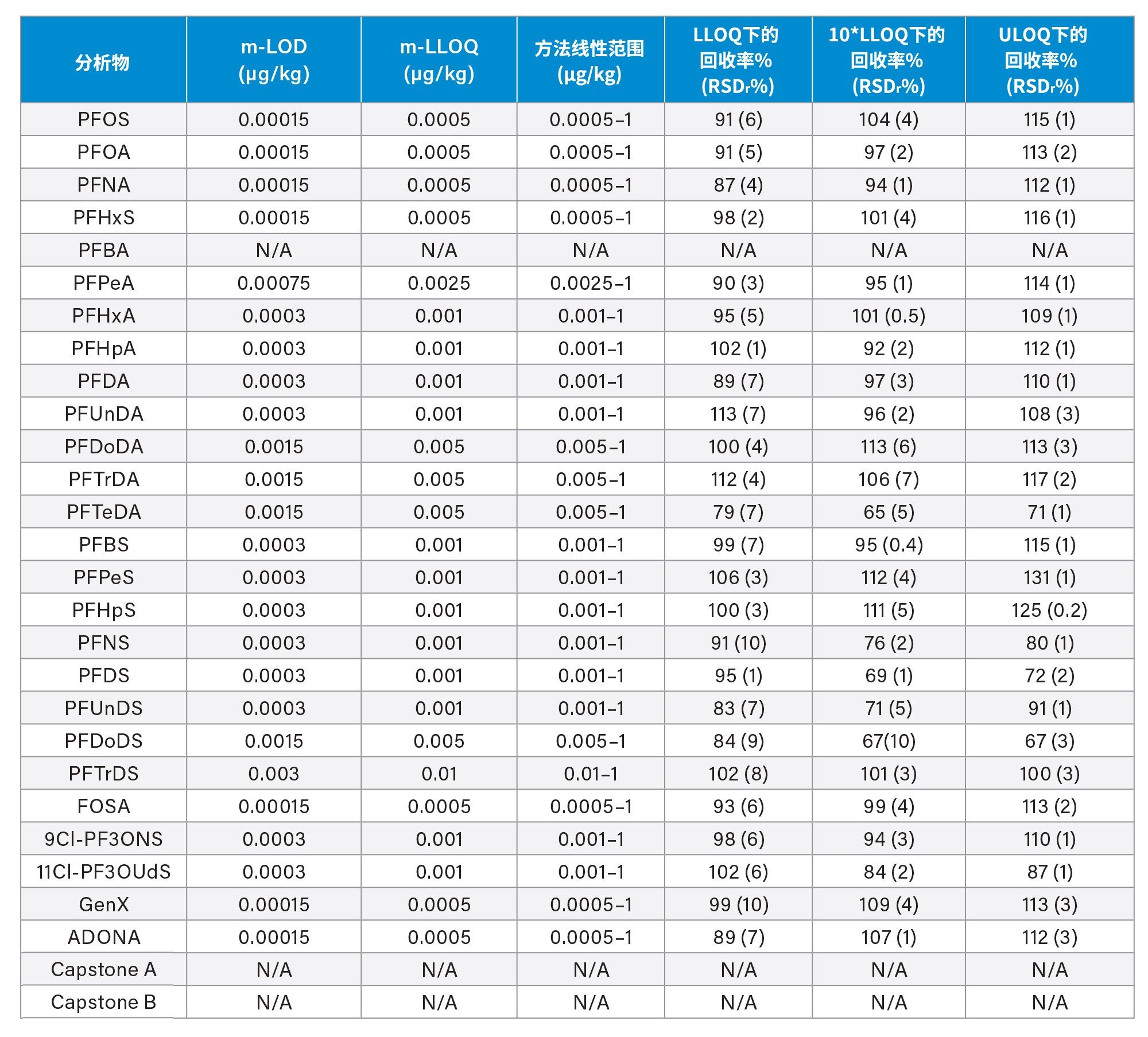
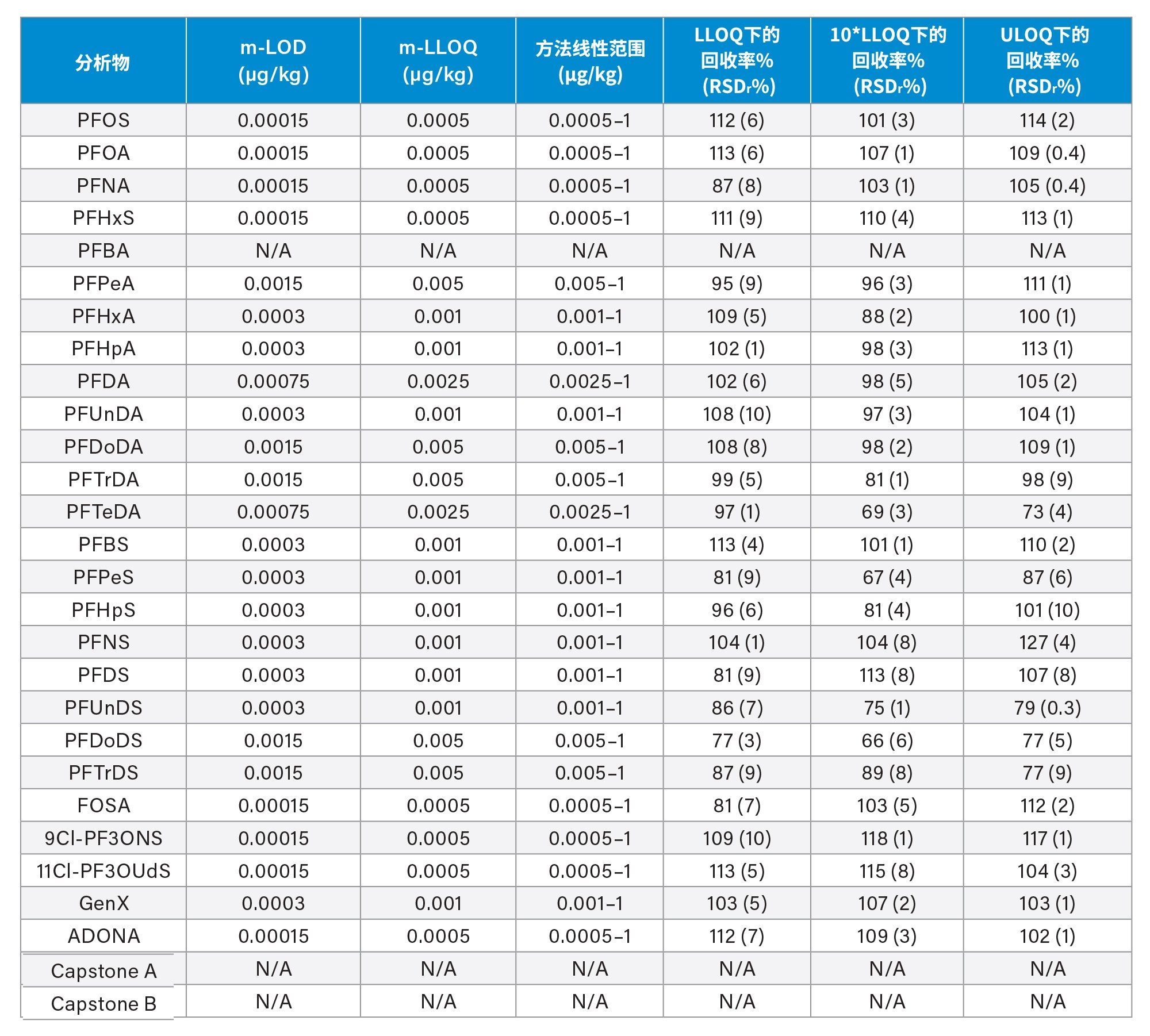
通过向空白样品中添加三个浓度水平标准品的回收率实验评估了正确度(根据表观回收率确定)。将整个天然PFAS组放在一起考虑时,所有加标浓度下,西红柿的平均回收率百分比为101%±14%(最小值=65%,最大值=131%),苹果为98%±13%(最小值=65%,最大值=131%),婴儿食品为99%±13%(最小值=66%,最大值=127%)。四种强制检测化合物(PFOS、PFOA、PFNA和PFHxS)的表观回收率在84%~116%之间。欧盟委员会(EU) 2022/1431号法规建议中推荐和考虑监测的其余PFAS的表观回收率在65%~131%之间。两组结果的正确度均符合CIR、EURL POPS指南和AOAC SMPR中描述的可接受标准。所有基质在所有加标浓度下的重复性(RSDr)范围为0.4%~10%,处于AOAC SMPR中规定的RSDr限值内*。唯一例外是PFDS、9Cl-PF3ONS和11Cl-PF3OUdS,它们在苹果和婴儿食品中的回收率符合标准,但在番茄中的回收率分别只有53%、52%和58%。然而,由于这三种化合物的RSDr远低于10%,因此可以应用回收率校正因子,因为精密度不会受到影响。回收率和RSDr报告见表2、3和4,绘图见图5、6和7。
(*)本研究未评估中间精密度(RSDR)。
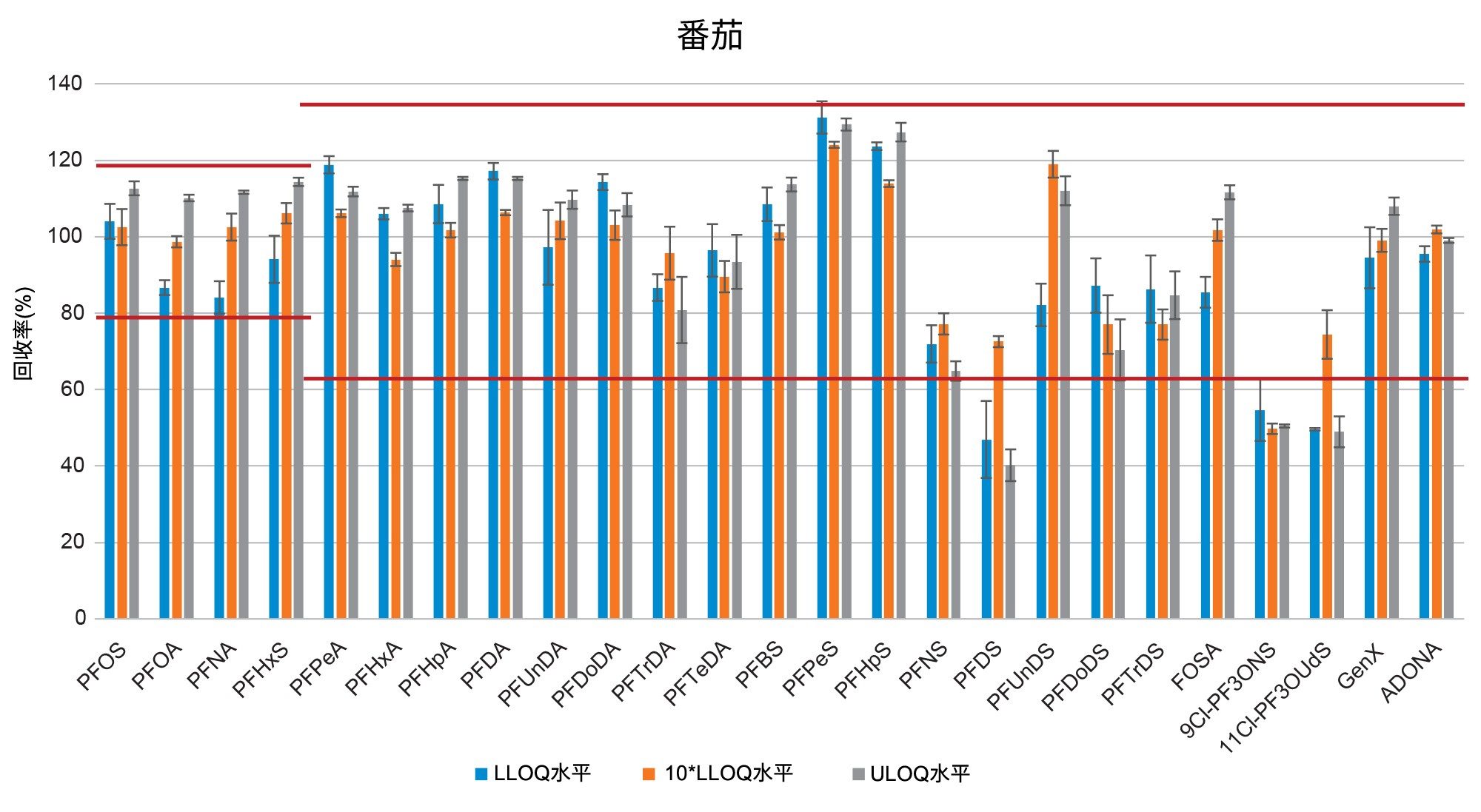
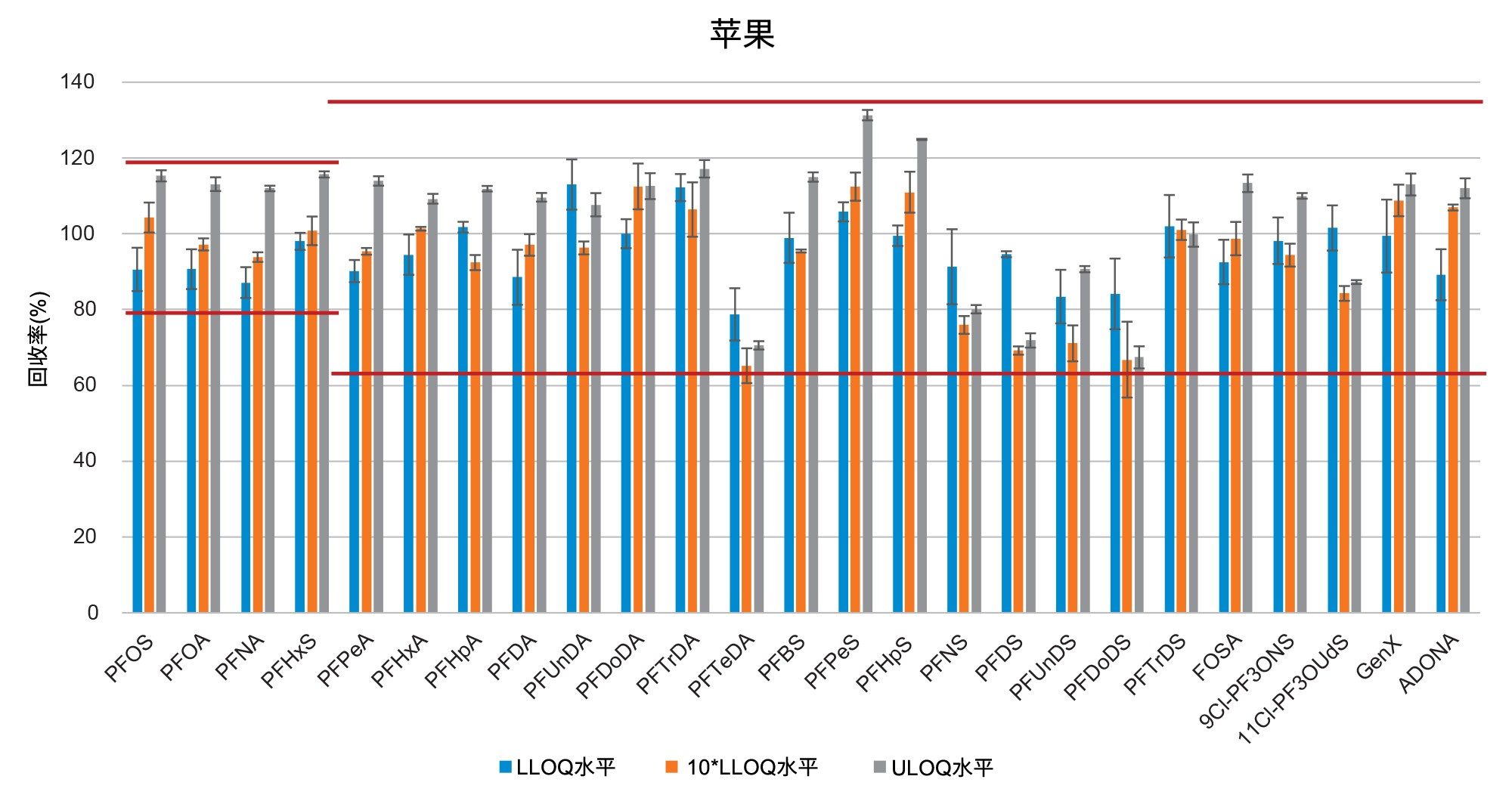
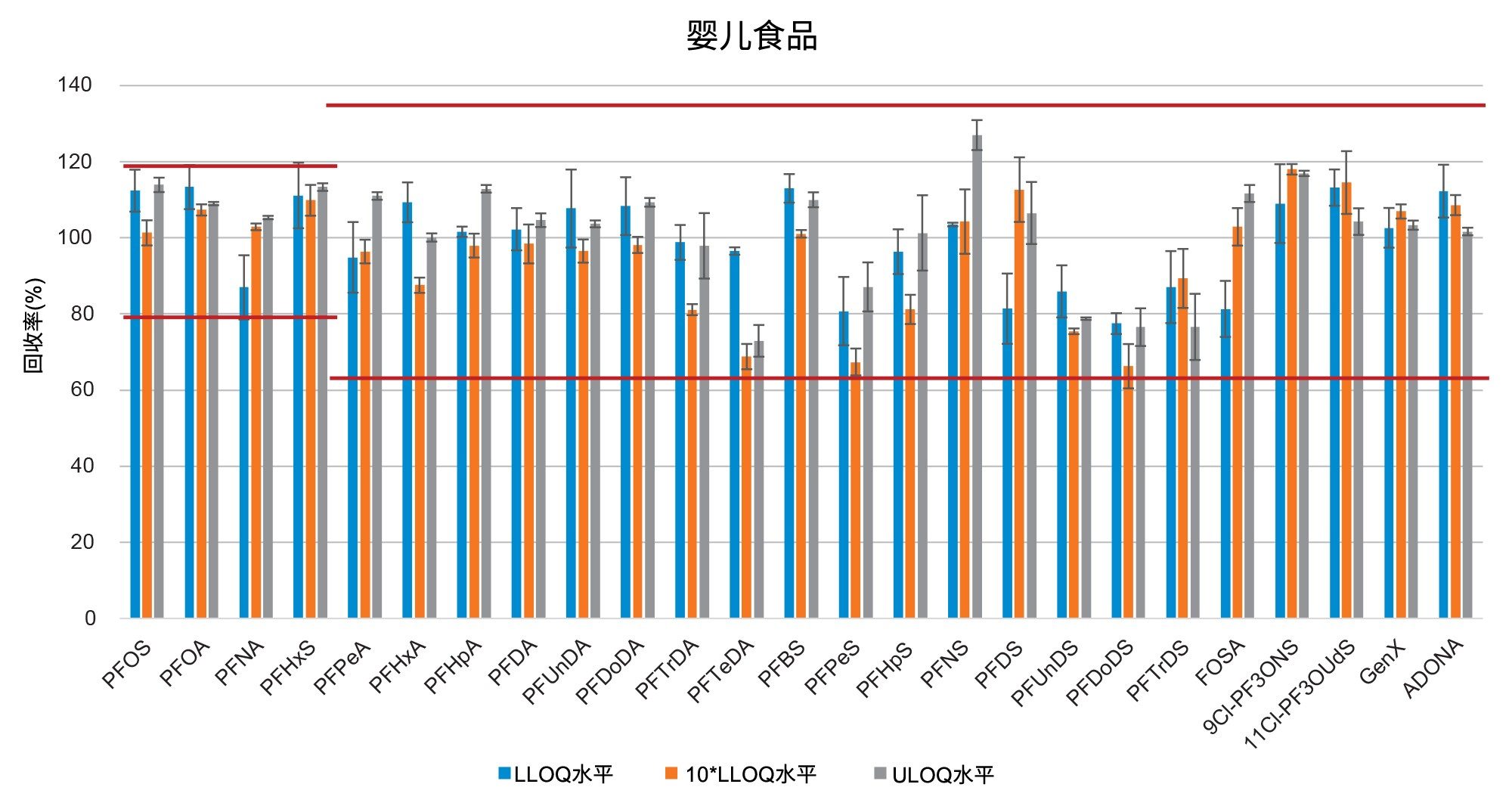
在本研究中,不仅基质样品中出现了非常高的PFBA污染,在溶剂和试剂(例如甲酸和甲醇)中也出现了同样的情况。在程序空白样品以及番茄和苹果中均检出PFBA,浓度为0.2 µg/kg。更令人担忧的是,在婴儿食品中也检出了PFBA,估计浓度为17.5 µg/kg(根据标准曲线推断)。PFBA污染因批次而异,但由于检测的所有类型和品牌的甲醇中都始终存在PFBA污染,因此在本研究中无法获得该化合物的准确回收率数据。
在空白基质中还检出了浓度接近m-LLOQ的许多PFAS化合物。在这种情况下,计算回收率时需要进行背景扣除。
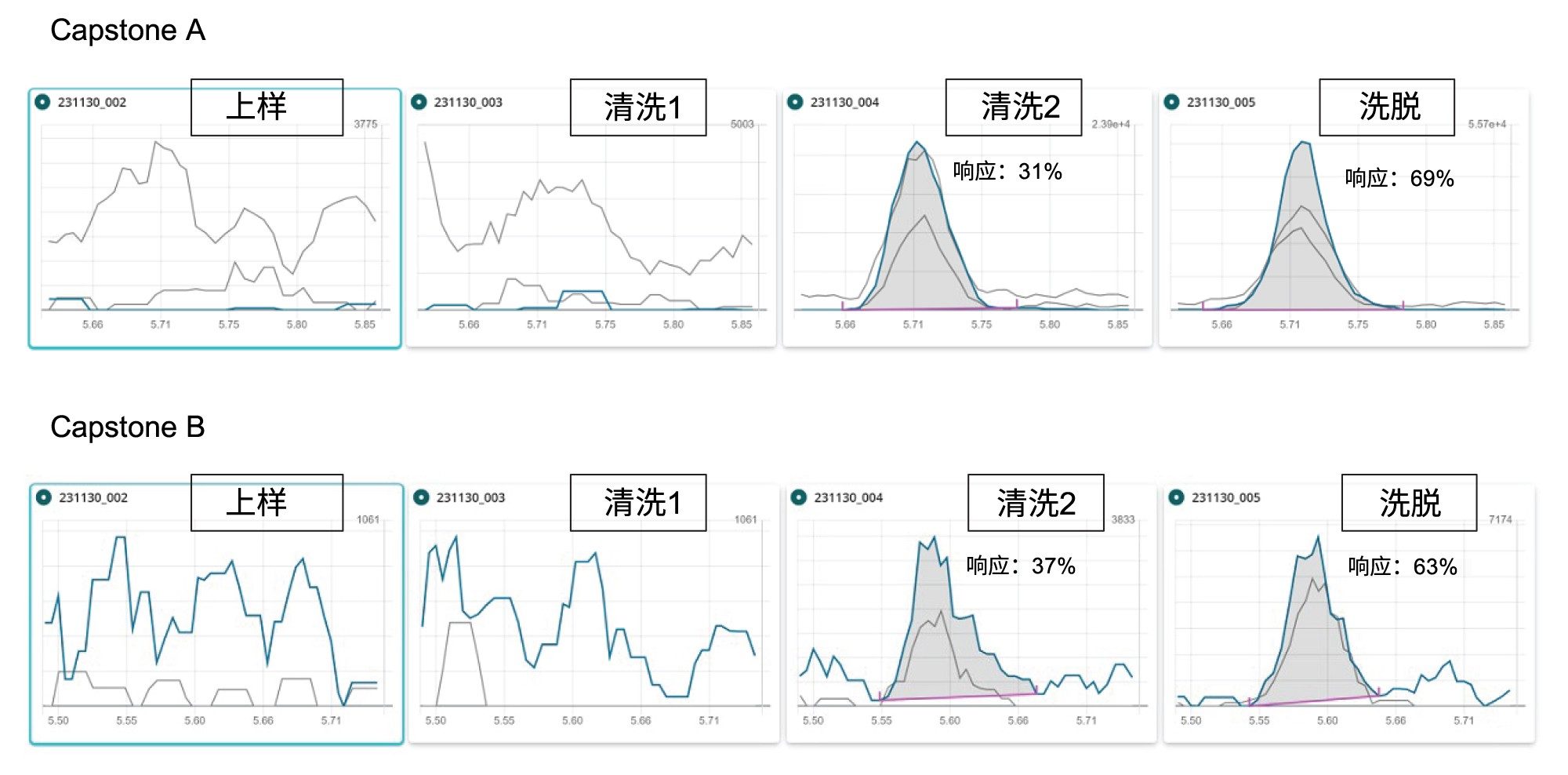
我们在方法开发阶段研究了Capstone A和B,其工艺回收率似乎非常低(低于10%)。为更好地了解其中原因,我们开展了一项实验,将0.5 ng/mL溶剂标准品上样至活化后的GCB/WAX小柱,收集SPE过程的每种馏分,用水或甲醇稀释,使样品瓶中的最终组成为1:1水:甲醇,然后进行分析。从图8可以看出,Capstone A和B在第二个清洗步骤中分别损失约31%和37%的分析物。一些研究人员11还发现,GCB/WAX SPE小柱无法有效保留这些两性离子化合物,因此他们建议将Capstone与其他PFAS分离,并通过“萃取-稀释-进样”法分析Capstone。导致这些化合物回收率较低的另一个因素可能是ESI中潜在的离子抑制现象。此外,由于Capstone A和Capstone B没有市售的标记内标,且化学结构特殊,为了实现更准确的定量分析,可以采用标准加入法。
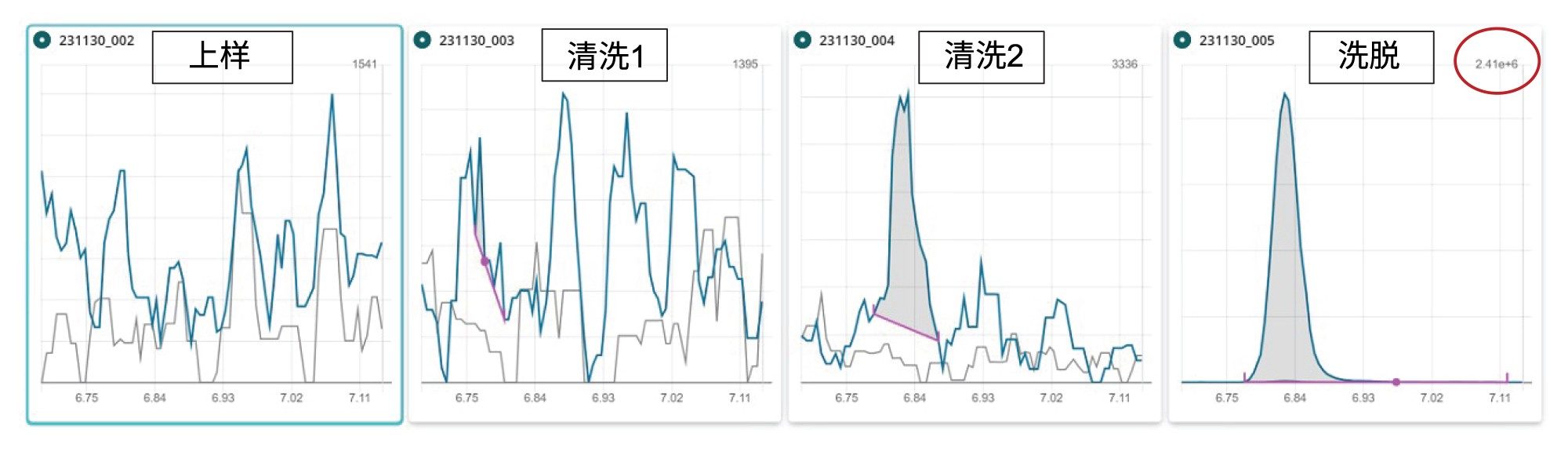
值得注意的是,FOSA的回收率在85%~112%范围内,表明本文所述的样品前处理和净化方法除适用于测定离子型PFAS以外,也适用于中性PFAS。我们对FOSA实施了与Capstone类似的实验,结果表明该化合物在洗脱液馏分中几乎完全回收(见图9)。
结论
本研究介绍了一种针对蔬菜、水果和婴儿食品中的PFAS进行全面分析的优化LC-MS/MS方法。该方法的定量限非常低(某些化合物可低至0.0005 µg/kg),能够准确检测和定量欧盟委员会(EU) 2022/1431号法规建议中列出的PFAS化合物。
净化效力更高的新型GCB/WAX SPE小柱,结合灵敏度增强的Xevo TQ Absolute MS系统,为强制检测的PFAS和大多数推荐检测和考虑检测的化合物提供了优异的回收率,分别为87%~116%和65%~131%,RSDr≤10%。
该方法还使FOSA(一种通常难以保留在弱阴离子交换小柱上的中性PFAS)获得了优异的回收率。
Capstone A和B的分析存在局限性,这是因为该化合物具有复杂的两性离子结构,以及溶剂和试剂存在PFBA污染。因此,我们需要开展更多研究,寻找既能减少PFBA污染,又能使Capstone获得更高回收率的理想方法。
本研究的第二部分将介绍动物产品中PFAS测定的工作流程。
参考资料
- Commission Regulation (EU) 2022/2388, amending Regulation (EC) No 1881/2006 as regards maximum levels of perfluoroalkyl substances in certain foodstuffs, L 316/38, 8.12.2022.
- Commission Recommendation (EU) 2022/1431, on the monitoring of perfluoroalkyl substances in food.L 221/105, 26.8.2022.
- Commission Implementing Regulation (EU) 2022/1428, laying down methods of sampling and analysis for the control of perfluoroalkyl substances in certain foodstuffs.L 221/66, 26.08.2022.
- EURL for halogenated POPs in feed and food (2022): Guidance Document on Analytical Parameters for the Determination of Per- and Polyfluoroalkyl Substances (PFAS) in Food and Feed, version 1.2 of 11 May 2022.在线获取:https://eurl-pops.eu/core-working-groups#_pfas.
- EURL for halogenated POPs in feed and food (2022): Guidance Document on Analytical Parameters for the Determination of Per- and Polyfluoroalkyl Substances (PFAS) in Food and Feed, ANNEX version 1.0 of 11 May 2022.在线获取:https://eurl-pops.eu/core-working-groups#_pfas.
- Standard Method Performance Requirements (SMPRs®) for Per- and Polyfluoroalkyl Substances (PFAS) in Produce, Beverages, Dairy Products, Eggs, Seafood, Meat Products, and Feed.AOAC SMPR® 2023.003. https://www.aoac.org/wp-content/uploads/2023/11/SMPR-2023_003.pdf.
- Dreolin N., Foddy H., Organtini K., Adams S., Rosnack K., Hancock P. (2023), Best practices for monitoring PFAS contamination in a routine shared-space commercial laboratory.Waters White Paper. 720007905.
- Organtini K., Oehrle S., Hird S., Adams S., Jandova R. (2021), 基质匹配还是同位素稀释?⽐较⽤于测定乳制品中PFAS的两种定量⽅法.沃特世应用纪要.720007687ZH.2022年8月.
- Adams S., Dreolin N., Organtini K., Hancock P. (2023).以鱼类参比样品作为案例研究,使用高灵敏度LC-MS/MS方法分析动物产品中的全氟烷基和多氟烷基化合物(PFAS).沃特世应用纪要.720008108ZH.2023年10月.
- Magnusson B. and Örnemark U. (eds.)Eurachem Guide: The Fitness for Purpose of Analytical Methods – A Laboratory Guide to Method Validation and Related Topics, (2nd ed.2014).ISBN 978-91-87461-59-0.
- Theurillat X., Mujahid C., Eriksen B., Griffin A., Savage A., Delatour T., Mottier P. (2023) An LC-MS/MS method for the quantitative determination of 57 per- and polyfluoroalkyl substances at ng/kg levels in different food matrices. Food Additives & Contaminants: Part A, 40:7, 862–877, DOI: 10.1080/19440049.2023.2226771.
附录
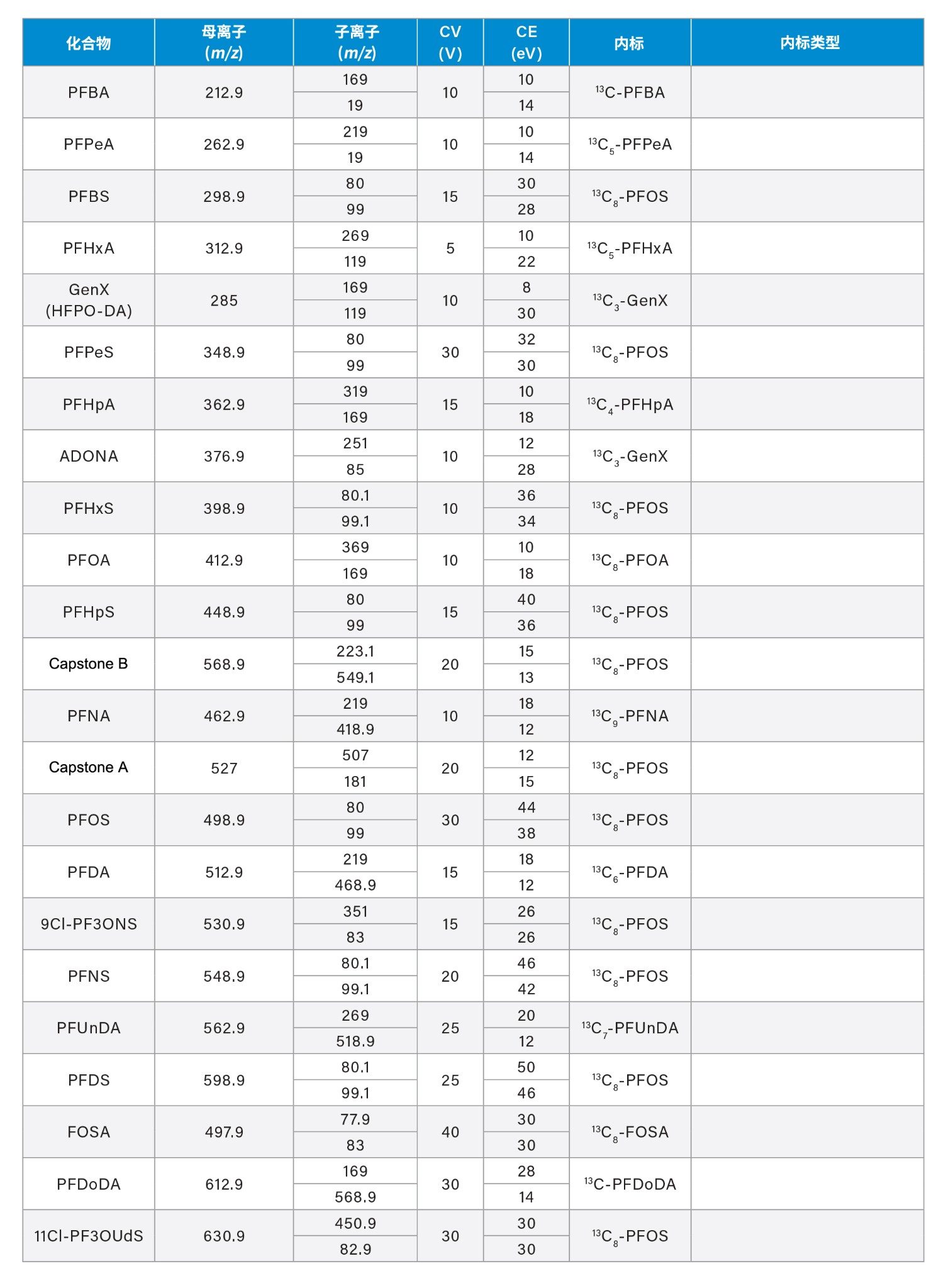
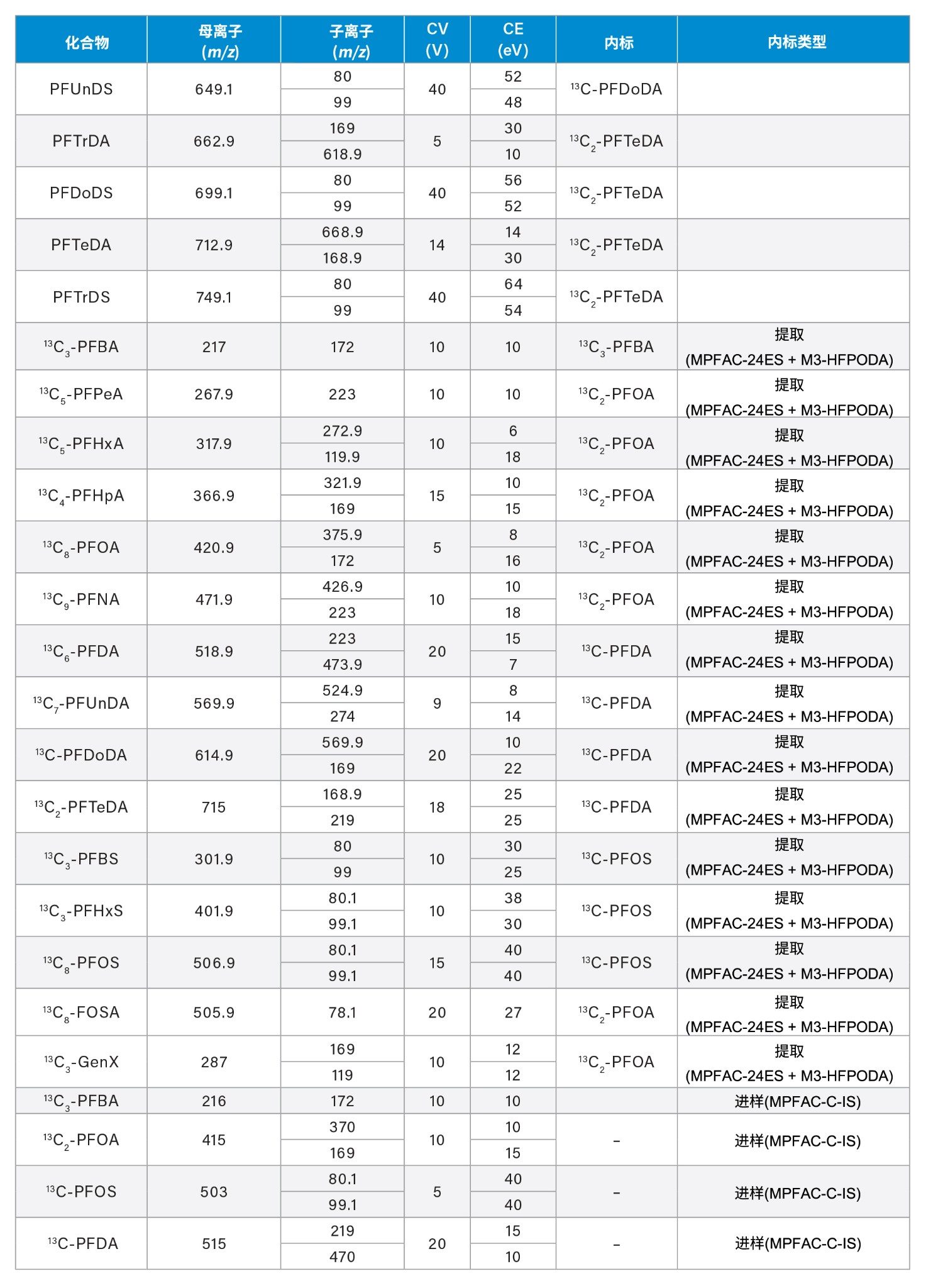
720008219ZH,2024年2月