利用Xevo™ TQ Absolute串联四极杆质谱仪和ACQUITY™ Premier系统对二甲双胍中的亚硝胺进行高灵敏度定量分析
摘要
药品中存在亚硝胺杂质会对人体健康构成极大风险,必须使用高灵敏度和选择性的分析方法进行监测,并要求精度达到亚ng/mL水平。本文主要介绍一种使用串联四极杆质谱仪开发的超高效液相色谱(UPLC™)方法用于检测和定量分析二甲双胍药物中九种亚硝胺杂质(NDMA、NDEA、NEIPA、NMOR、NDIPA、NDPA、NMPA、NMBA、NDBA)的性能。使用Atlantis™ Premier™ BEH C18 AX色谱柱进行色谱分离,使用ACQUITY Premier系统和Waters Xevo TQ Absolute质谱仪进行灵敏且准确的定量分析。亚硝胺在纯溶剂和二甲双胍药物中的定量限(LOQ)分别为0.01~0.1ng/mL和0.025~0.1ng/mL。本研究所用方法在0.025 ng/mL(或者0.00125 ppm,相对于20 mg/mL二甲双胍药物)时实现准确的定量性能,回收率为85~110%。
优势
- 使用Xevo TQ Absolute串联四极杆质谱仪在MRM采集模式下对二甲双胍药物中的亚硝胺进行痕量检测
- 使用Atlantis Premier BEH C18 AX色谱柱搭配ACQUITY Premier系统可靠地分离二甲双胍药物中的亚硝胺
- 对二甲双胍药物中的亚硝胺定量性能好,精密度、重复性、线性和准确度均出色
简介
亚硝胺杂质被视为潜在的人类致癌物或可致癌的化合物1,2。自2018年起,由于在几款市售药物中发现亚硝胺,导致产品召回,继而引起市场混乱。这些药物包括血管紧张素II受体阻滞剂(ARBs)、雷尼替丁抗组胺药(Zantac)以及之后的二甲双胍3,4。 为控管药品中存在的亚硝胺,美国FDA和欧洲药品管理局(EMA)规定了药品中亚硝胺可接受的每日摄入限量(纳克/天)4,5。 根据特定药品的推荐每日最大剂量,使用这些限值确定药品中亚硝胺的阈值浓度。
二甲双胍是一种处方药,用于治疗2型糖尿病患者的高血糖3。 有几款二甲双胍药品由于NDMA含量超过了96纳克/天的可接受摄入限值而被召回3。
要测量法规允许的阈值水平的亚硝胺,需要使用高灵敏度和选择性的分析方法。液质联用(LC-MS)仪器已成功应用于各种药品中微量亚硝胺的准确鉴别和定量6,7。
本文所述的UPLC-MS/MS方法对二甲双胍药物或药物活性成分(API)中的九种亚硝胺(表1)进行了高灵敏度和选择性的检测和定量分析。该方法使用Xevo TQ Absolute串联四极杆质谱仪和ACQUITY Premier系统开发。本文展示了方法的性能特征,包括二甲双胍药物的检测限(LOD)和定量限(LOQ)、重现性、线性以及准确度。
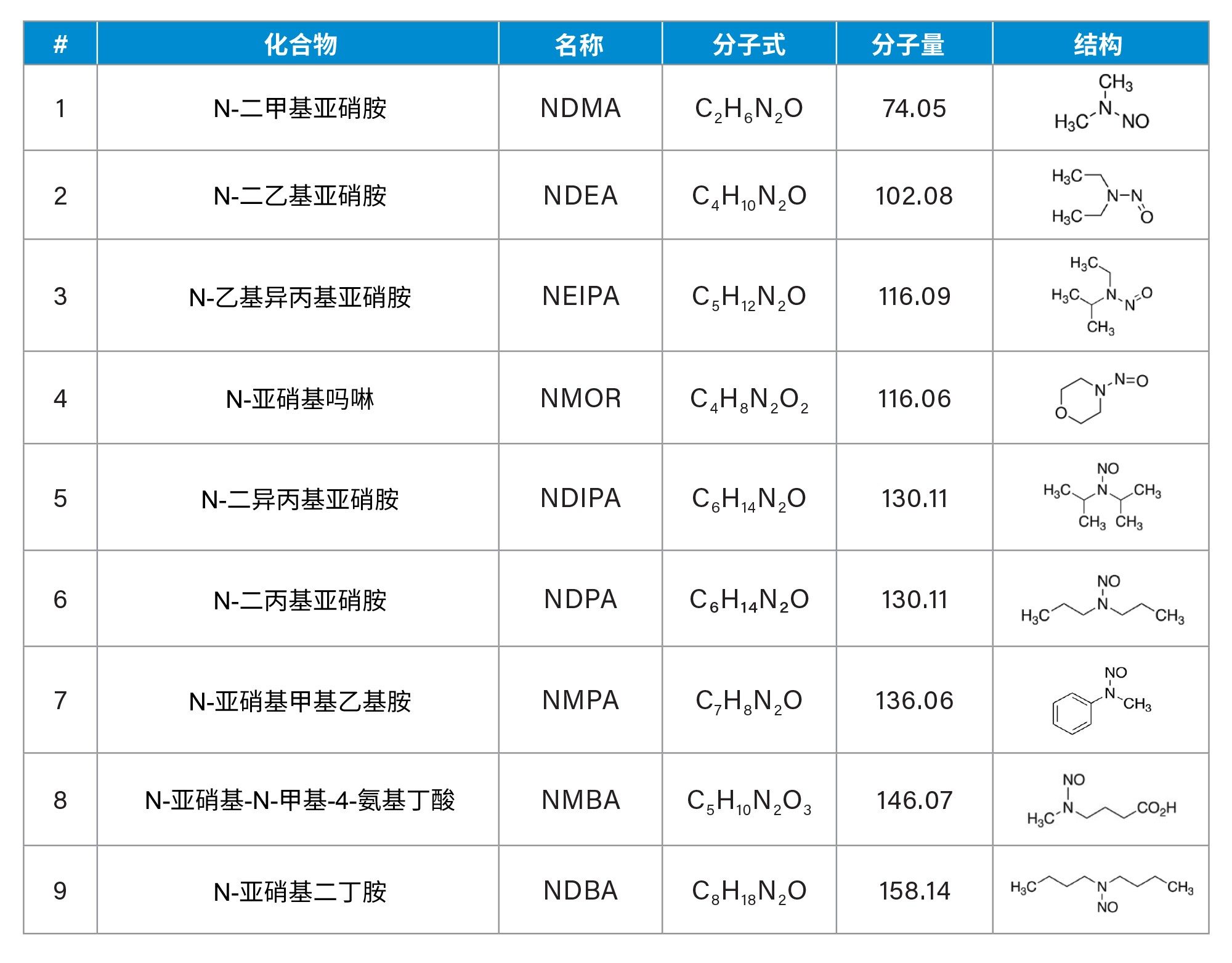
实验
亚硝胺标准品购自Toronto Research Chemicals (TRC)和Sigma-Aldrich。质谱级甲酸铵、溶剂和甲酸购自Honeywell。二甲双胍药物购自Sigma-Aldrich。
用纯溶剂制备的标准溶液
将每种亚硝胺各5.0-10 mg/mL的单标储备液溶于甲醇中,制备含九种亚硝胺(100 µg/mL)的混标溶液。用水连续稀释混标溶液,制备LOD、LOQ和线性标准溶液。
二甲双胍药物(DS)
用水制备浓度为20 mg/mL的二甲双胍药物样品溶液,然后用0.2 µm/mL的PVDF针头式过滤器(P/N: WAT200806)进行过滤以备分析
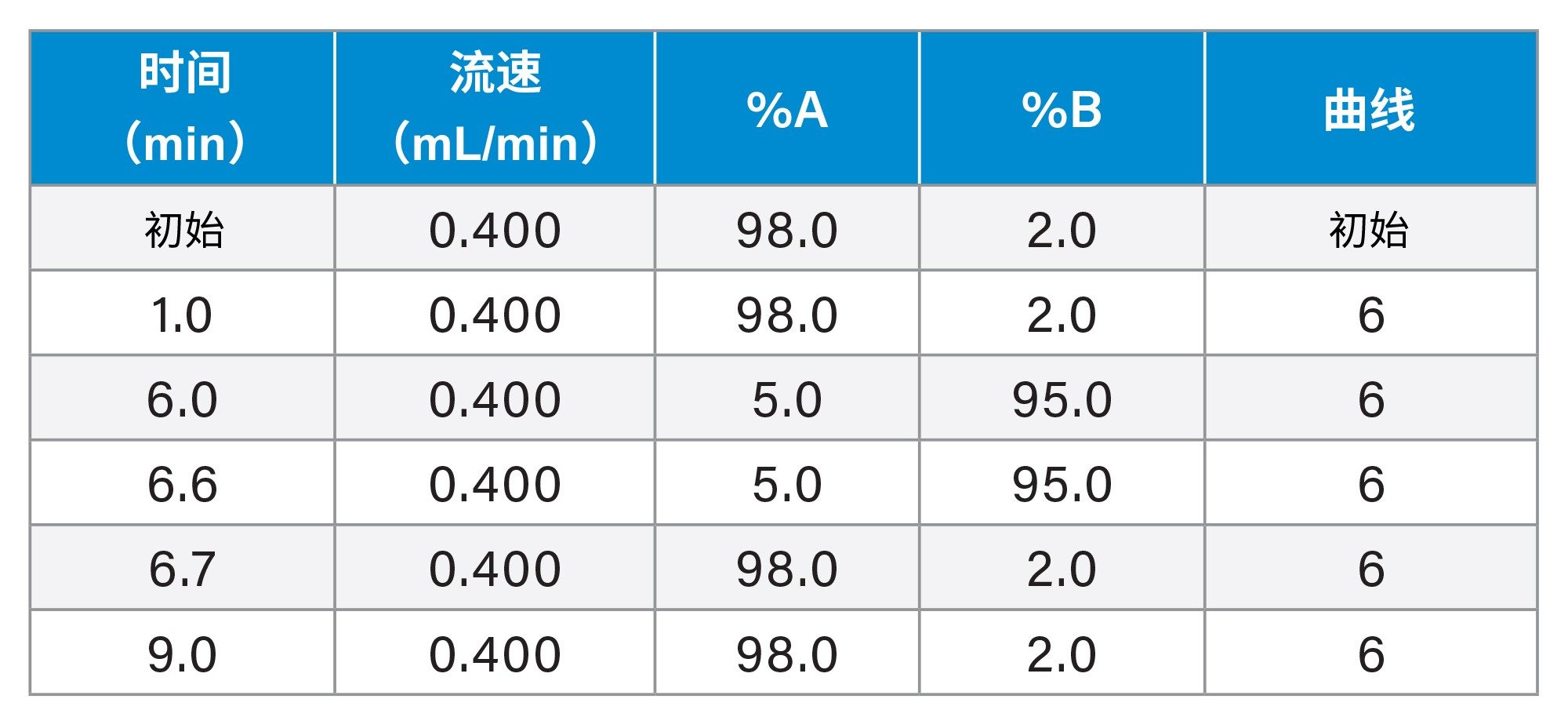
液相色谱条件
|
液相色谱系统: |
ACQUITY™ Premier系统 |
|
检测条件: |
MS/MS |
|
样品瓶: |
LCMS最大回收样品瓶,容积2 mL,P/N:600000670CV |
|
色谱柱: |
Atlantis™ Premier BEH C18 AX (2.1 x 100, 1.7 um),P/N:186009368 |
|
柱温: |
40°C |
|
样品温度: |
10°C |
|
进样体积: |
30.0 µL |
|
流速: |
0.4 mL/min |
|
流动相A: |
含0.1%甲酸和5 mM甲酸铵的水溶液 |
|
流动相B: |
含0.1%甲酸和5 mM甲酸铵的甲醇溶液 |
|
梯度: |
见梯度表 |
梯度表
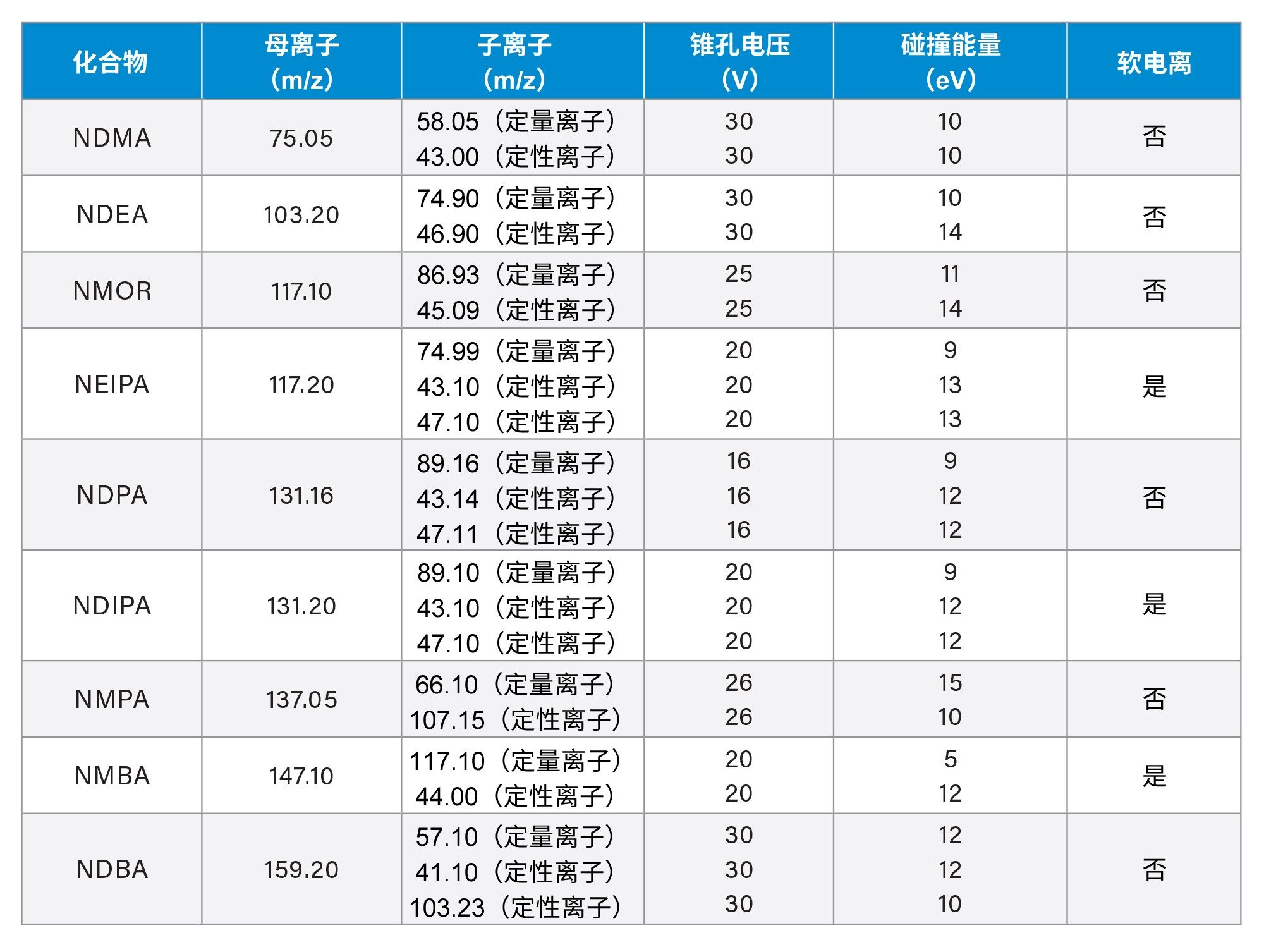
质谱条件
|
质谱系统: |
Xevo™ TQ Absolute串联四极杆质谱仪 |
|
电离模式: |
APCI+ |
|
采集: |
MRM模式,如表2所述 |
|
电晕针电流: |
2.5 (µA) |
|
APCI探头温度: |
325°C |
|
脱溶剂气流速: |
950 L/h |
|
锥孔气流速: |
300 L/h |
|
喷雾器: |
300 L/h |
|
碰撞气体流速: |
0.20 mL/min |
|
离子源温度: |
150°C |
数据管理
|
数据管理: |
仪器控制:MassLynx™ v4.2 |
|
数据处理:TargetLynx™ |
结果与讨论
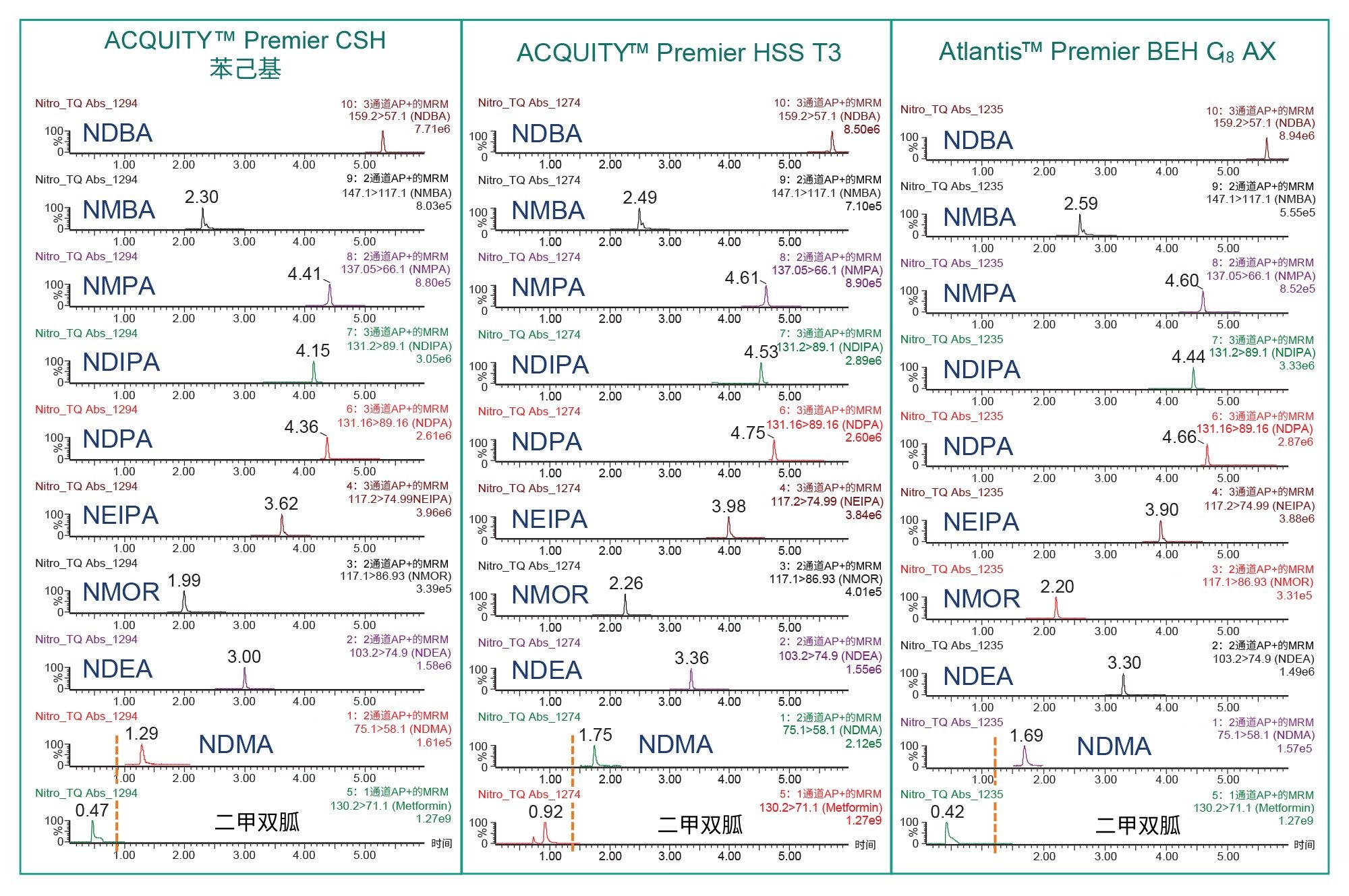
方法开发过程中考察了各种色谱柱填料,确保所有亚硝胺都能够实现色谱分离,尤其是分离高浓度二甲双胍API峰和强极性的NDMA峰。确保分离高浓度API峰和微量杂质,使集成转换阀将API导向废液,同时将杂质导向MS进行分析。将API导向废液可以尽量减少基质可能对痕量杂质的离子抑制或增强造成的影响。
亚硝胺和二甲双胍分析的色谱柱筛选结果如图1所示。为了评估二甲双胍峰的洗脱,使用含10 ng/mL二甲双胍药物以及1 ng/mL亚硝胺的样品,在不用转换阀的情况下采集MS迹线。ACQUITY Premier HSS T3和ACQUITY CSH™苯己基柱为所有亚硝胺提供了足够的保留时间,而ACQUITY Atlantis Premier BEH C18 AX使二甲双胍和NDMA获得了理想的色谱分离效果(图1)。此外,BEH C18 AX色谱柱提高了邻近洗脱的亚硝胺(NDPA、NDIPA和NMPA)之间的分离度。
根据以前分析六种亚硝胺(NDMA、NDEA、NEIPA、NDIPA、NDBA和NMBA8)的结果,本研究使用大气压化学电离(APCI)在正离子模式下对亚硝胺进行检测和定量分析。使用MassLynx软件中的IntelliStart™功能开发亚硝胺的MRM通道和MS电离参数。
纯溶剂中的定量分析
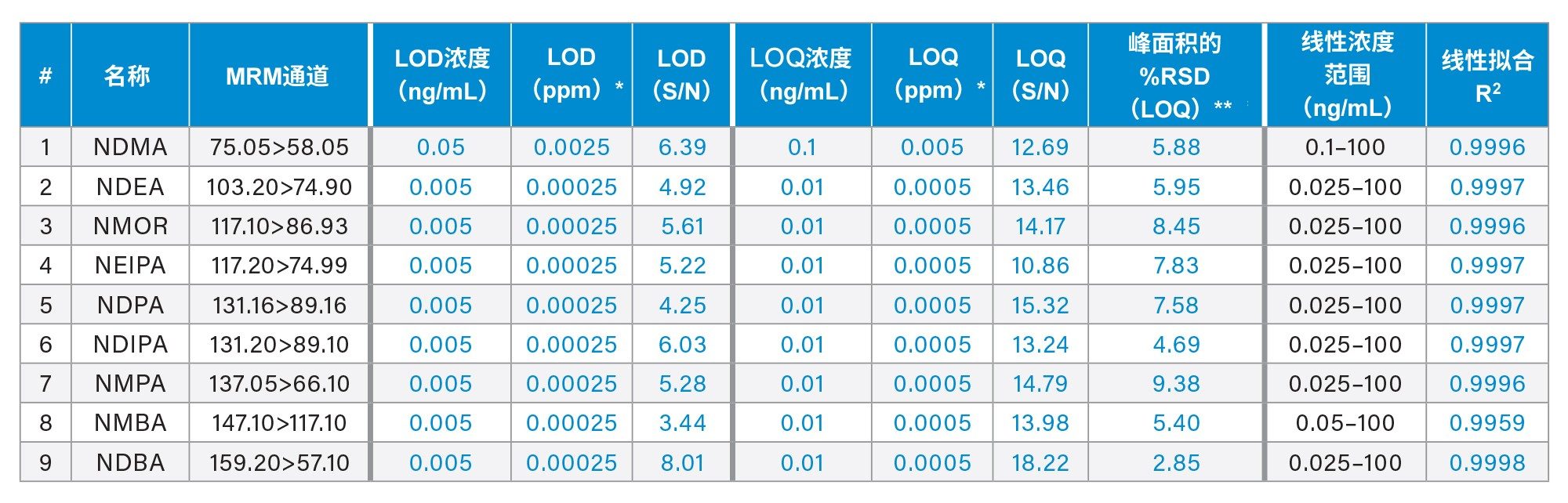
该UPLC-MS/MS方法所能达到的亚硝胺LOD和LOQ分别是按照3:1和10:1的信噪比(S/N)标准确定的。纯溶剂中LOQ溶液的代表性色谱图如图2所示。表3总结了方法的性能特征,包括LOD、LOQ和线性。NDMA的LOD和LOQ分别为0.05 ng/mL和0.1 ng/mL。此外,NDEA、NMOR、NEIPA、NDPA、NDIPA、NMPA、NMBA和NDBA的LOD和LOQ分别为0.005 ng/mL和0.01 ng/mL。0.1和0.01 ng/mL的LOQ对于20 mg/mL二甲双胍药物来说相当于0.005和0.0005 ppm。根据六次重复进样的数据(表3),峰面积的相对标准偏差(RSD)低于10%,证明该方法对于亚硝胺的LOQ方面性能优异。本研究没有使用内标来校正数据差异性。此外,该方法显示了MS响应和浓度之间的线性关系,相关系数≥0.996(表3)。
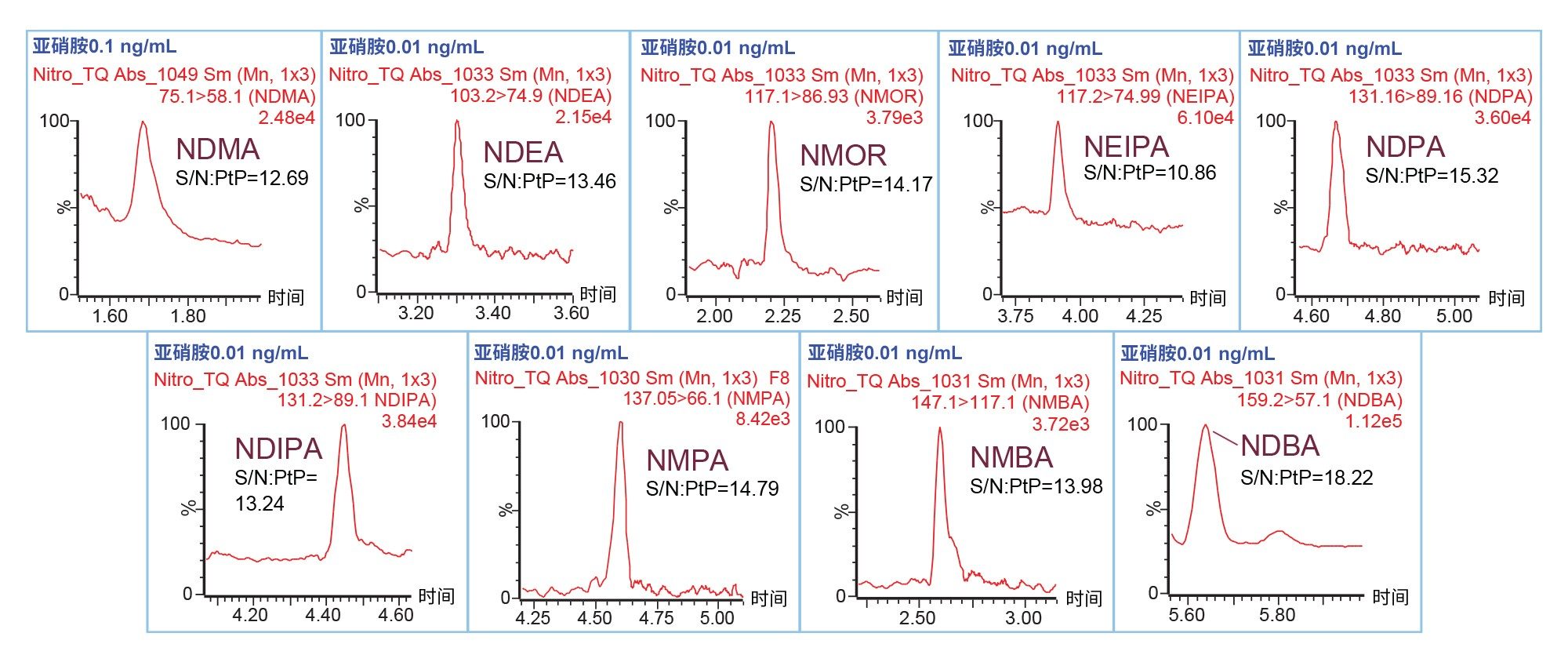
二甲双胍药物中的定量分析
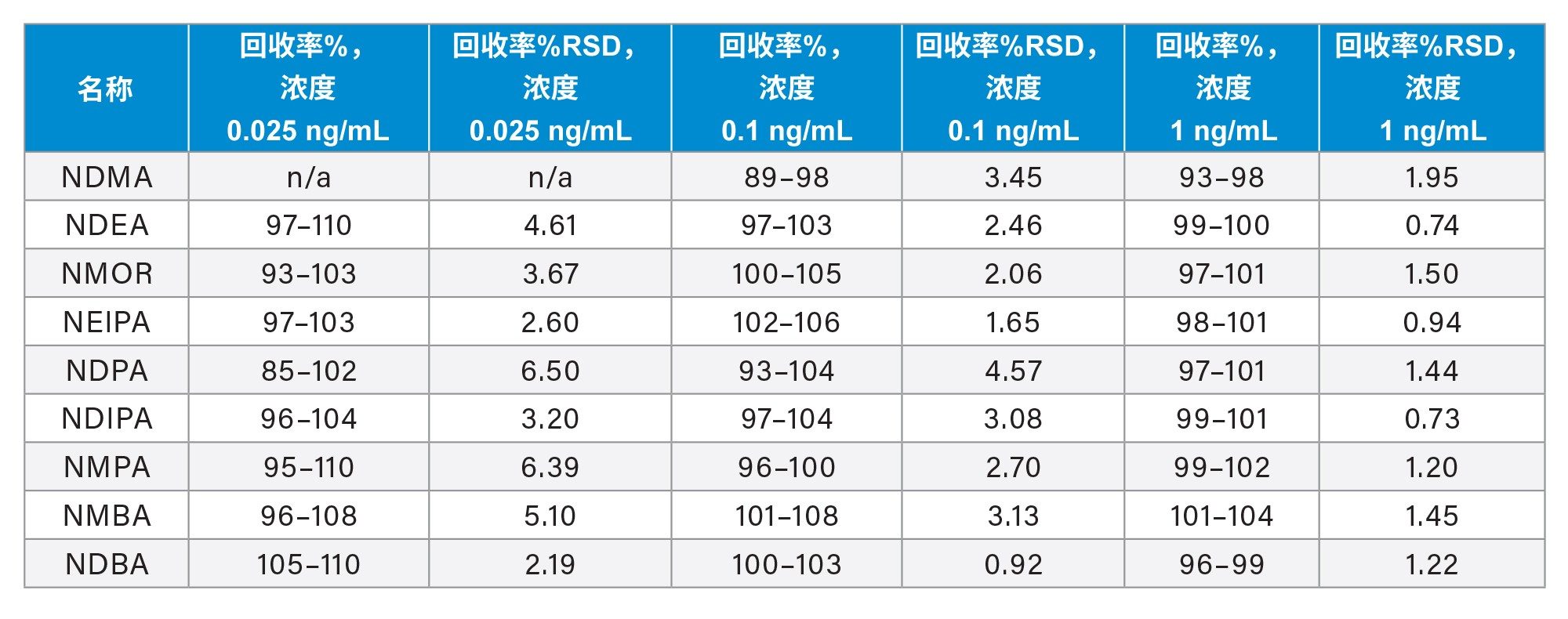
在MRM采集模式下分析20 mg/mL的二甲双胍药物水溶液,检测其中存在的亚硝胺。为了评估方法准确度,向二甲双胍样品中分别加入0.025、0.1和1 ng/mL亚硝胺。此操作证明,该方法可以准确测量高浓度二甲双胍供试品中的亚硝胺杂质。分析二甲双胍药物和加标后二甲双胍药物样品中亚硝胺(NDMA、NDEA、NMOR和NEIPA)的代表性色谱图如图3所示。分析结果证明,本研究检测的20 mg/mL二甲双胍药物中未检出亚硝胺。此外,二甲双胍加标样品中亚硝胺的MS响应和S/N与纯标准溶液中的值相当,表明高浓度二甲双胍峰没有离子抑制作用。
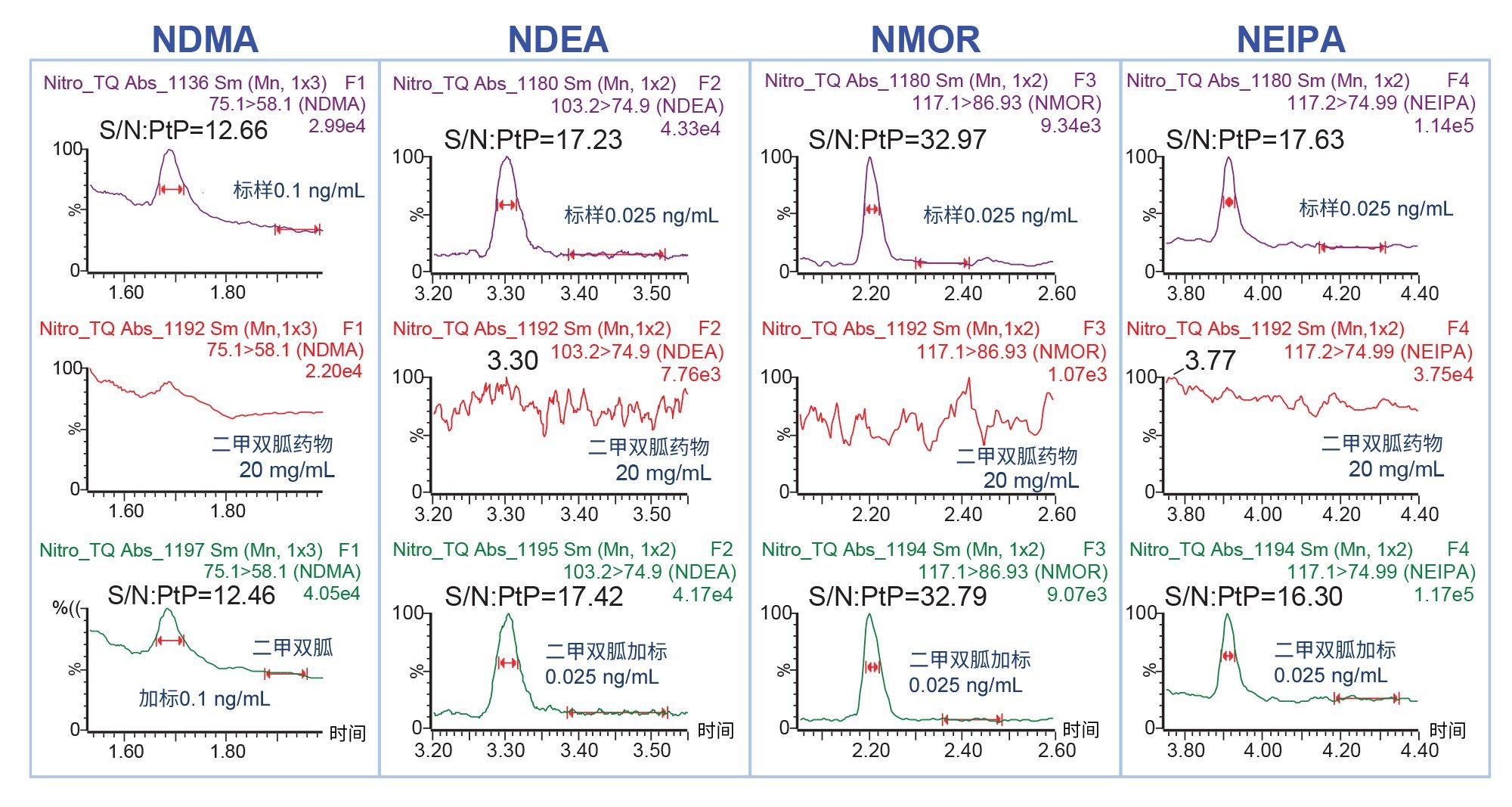
使用TargetLynx软件,通过对比计算浓度和已知的加标浓度计算出百分比回收率。用二甲双胍药物制备的校准标样计算亚硝胺的回收率。为了准确定量,一般建议使用与样品基质相同的标样。如果基质中含有分析物或干扰物,可以使用标准加入法。
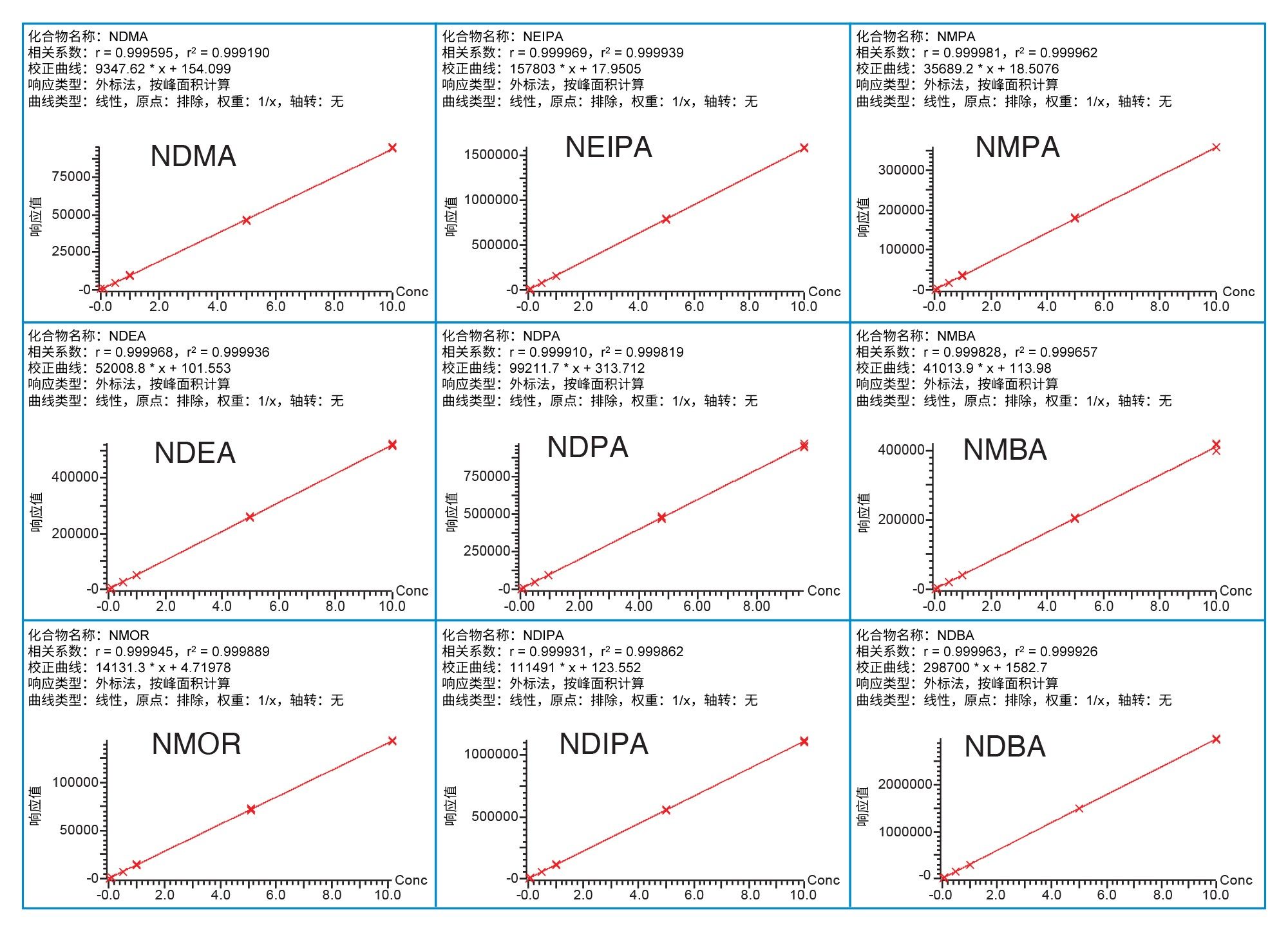
二甲双胍药物中亚硝胺标准品的标准曲线显示MS响应与浓度呈线性关系,R2≥0.999(图4)。该方法对二甲双胍药物中的所有亚硝胺均表现出优异的准确度(表4)。对于NDMA,0.1 ng/mL和1 ng/mL时的回收率分别为89~98%和93~98%。对于其他杂质,0.025 ng/mL(或0.00125 ppm,相对于20 mg/mL二甲双胍药物)时的回收率为85~110%,5个样品的RSD ≤ 6.50%。
结论
本研究利用Xevo TQ Absolute质谱仪和ACQUITY Premier系统开发了一种高灵敏度方法,能够对二甲双胍药物中的亚硝胺进行超微量检测和定量分析。使用Atlantis™ Premier BEH C18 AX色谱柱完成出色的色谱分离。该方法显示出优异的定量性能,在纯溶剂和20 mg/mL二甲双胍药物中的LOQ分别达到0.01~0.1 ng/mL和0.025~0.1 ng/mL。二甲双胍药物中亚硝胺的线性和准确度结果分别为R2 ≥ 0.999、回收率85~110%。
本文所述的UPLC-MS/MS方法可对二甲双胍药物中的亚硝胺进行高灵敏度、高专属性和准确的分析,达到准确监测残留亚硝胺的目的,这对于产品质量和安全至关重要。
参考资料
- ICH M7(R1) Assessment and Control of NDMA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk, International Conference on Harmonization.
- G. Brambilla, A. Martelli, Genotoxic and Carcinogenic Risk to Humans of Drug–Nitrite Interaction Products, Mutat.Res.635 (2007) 17–52.
- https://www.fda.gov/news-events/press-announcements/fda-alerts-patients-and-health-care-professionals-nitrosamine-impurity-findings-certain-metformin.
- FDA, Control of Nitrosamine Impurities in Human Drugs Guidance for Industry, U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER), February 2021.
- European Medicines Agency, Nitrosamine impurities in human medicinal products, Procedure number: EMEA/H/A-5(3)/1490, 25 June 2020.
- Parr MK and Joseph JF.NDMA impurity in valsartan and other pharmaceutical products: Analytical methods for the determination of N-nitrosamines.JPBA 2018;164(2019):536–549.
- Gushargi AJ and Halden RU.Critical Review of Major Sources of Human Exposure to N-nitrosamines.Chemosphere 2018;210:1124-1136.
- Lame ME, Lindsay H. High sensitivity quantification of Nitrosamines Genotoxic Impurities: LC-MS Analysis of Ranitidine Drug Product Using the Waters ACQUITY UPLC I-Class/Xevo TQ-XS Tandem Quadrupole Mass Spectrometer.Waters Application Note, 720006899, 2020.
720007725ZH,2022年9月