使用液相色谱-高分辨率质谱法筛查萃取物的分析注意事项
摘要
随着全球法规日益严苛,因此对药品包装、食品接触材料、医疗器械和其他不同行业耗材中潜在萃取物和可浸出物(E&L)的成分进行筛查变得越来越重要。为了确保消费者安全并降低此类成分相关风险,需要对所有潜在有害化合物进行鉴定。
液相色谱-高分辨率质谱(LC-HRMS)是分析非挥发性有机化合物的常用技术。所用筛查方法需要能够对多种化学物质进行筛查,并且能够灵敏地筛查出此类低含量化合物。虽然目前已有法规对样品萃取技术提供了一些指导,但在开发分析仪器相关筛查方法方面目前并没有相关规定。分析人员需要自己设计和实施LC-HRMS筛查方法,以分析是否存在潜在E&L化合物。
本研究的目的是评估现有E&L筛查方法及其对典型E&L化合物的影响,以及首程通用筛查方法适用的方法条件组合。另外,还阐明了在就萃取物和可浸出物分析设计LC-HRMS筛查方法时需要考虑的一些额外因素。
优势
- 为初次设计用于萃取物和可浸出物筛查应用的通用LC-HRMS方法提供了提示和注意事项
- 灵敏的首程萃取物筛查方法,可对多种化学物质进行筛查
- 简单的液相色谱-高分辨率质谱联用方法,适用于筛查化妆品、食品接触材料、药品包装和医疗器械中的萃取物
简介
随着全球法规日益严苛,因此对药品包装、食品接触材料、医疗器械和其他不同行业耗材中潜在萃取物和可浸出物(E&L)的成分进行筛查变得越来越重要1,2。 为了确保消费者安全并降低此类成分相关风险,需要对存在的所有潜在有害化合物进行鉴定。
纵览这些成分的生产过程,从原料到最终产品,需要经过多个环节,其中每个环节都会引入额外的杂质和降解产物,由此可能会产生E&L。这些成分可能非常复杂,并且可能包含多种化学物质,因此需要使用多种分析技术进行筛查和表征。液相色谱-高分辨率质谱(LC-HRMS)是分析非挥发性有机化合物的常用技术。所用筛查方法需要能够对多种化学物质进行筛查,并且能够灵敏地筛查出此类低含量化合物。
虽然目前已有法规对样品萃取技术提供了一些指导,但在开发分析仪器相关筛查方法方面目前并没有相关规定3,4,5。分析人员需要自己设计和实施LC-HRMS筛查方法,以分析是否存在潜在E&L化合物。
本研究的目的是评估现有内部E&L筛查方法及其对典型E&L化合物的影响,以及首程通用筛查方法适用的方法条件组合。本文所述方法并不是优化方法,而是基于一系列现有E&L筛查方法使用数量有限的溶剂、添加剂和方法条件所开发的方法。此方法旨在帮助找到通用E&L筛查方法的最佳内部解决方案,从而用于对大量分析物和化学物质(目标分析物列表不一定已知)进行筛查。对于具有特定化学性质的一组特定分析物,建议采用更有针对性的优化方法。另外,还阐明了在就E&L分析设计LC-MS筛查方法时需要考虑的一些额外因素。
实验
分析方案
按照包含酸洗、碱洗和IPA清洗的全面清洗方案对系统进行了清洗,以期在标准品进样之前尽量减少污染和背景噪音。为了对不同类型的化合物进行评估,分别进样两种混标,即Waters LC-MS QC参比标准品(LC-MS混合物)和Waters E&L筛查标准品 (E&L混合物),以涵盖多种化合物极性、质量数,以及化学成分(在ESI(电喷雾电离)负离子和正离子模式下均可进行电离)(表1和表2)。在正离子和负离子模式下,均先运行甲醇空白进样,然后再进样标准品,每种标准品重复进样三次。
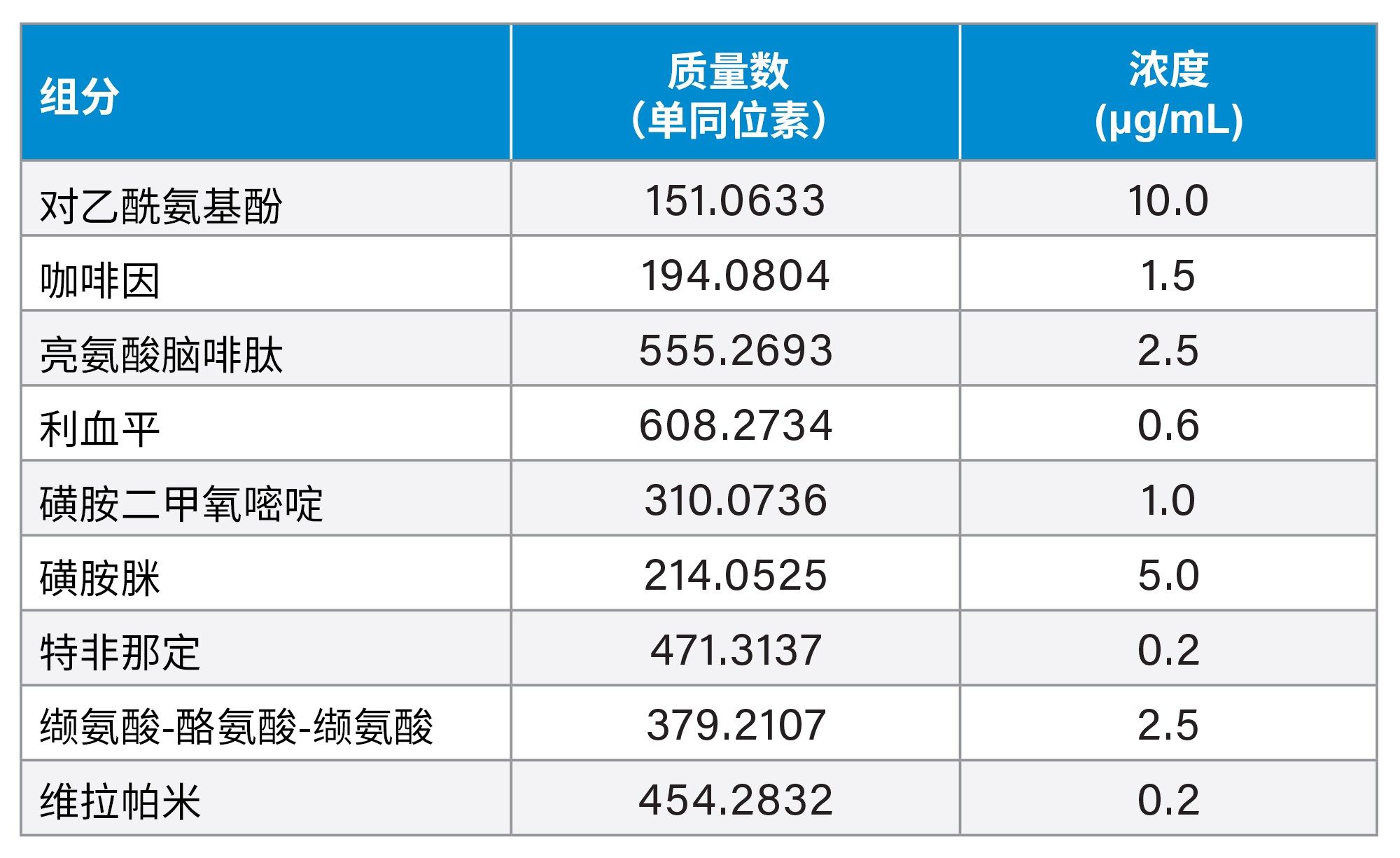
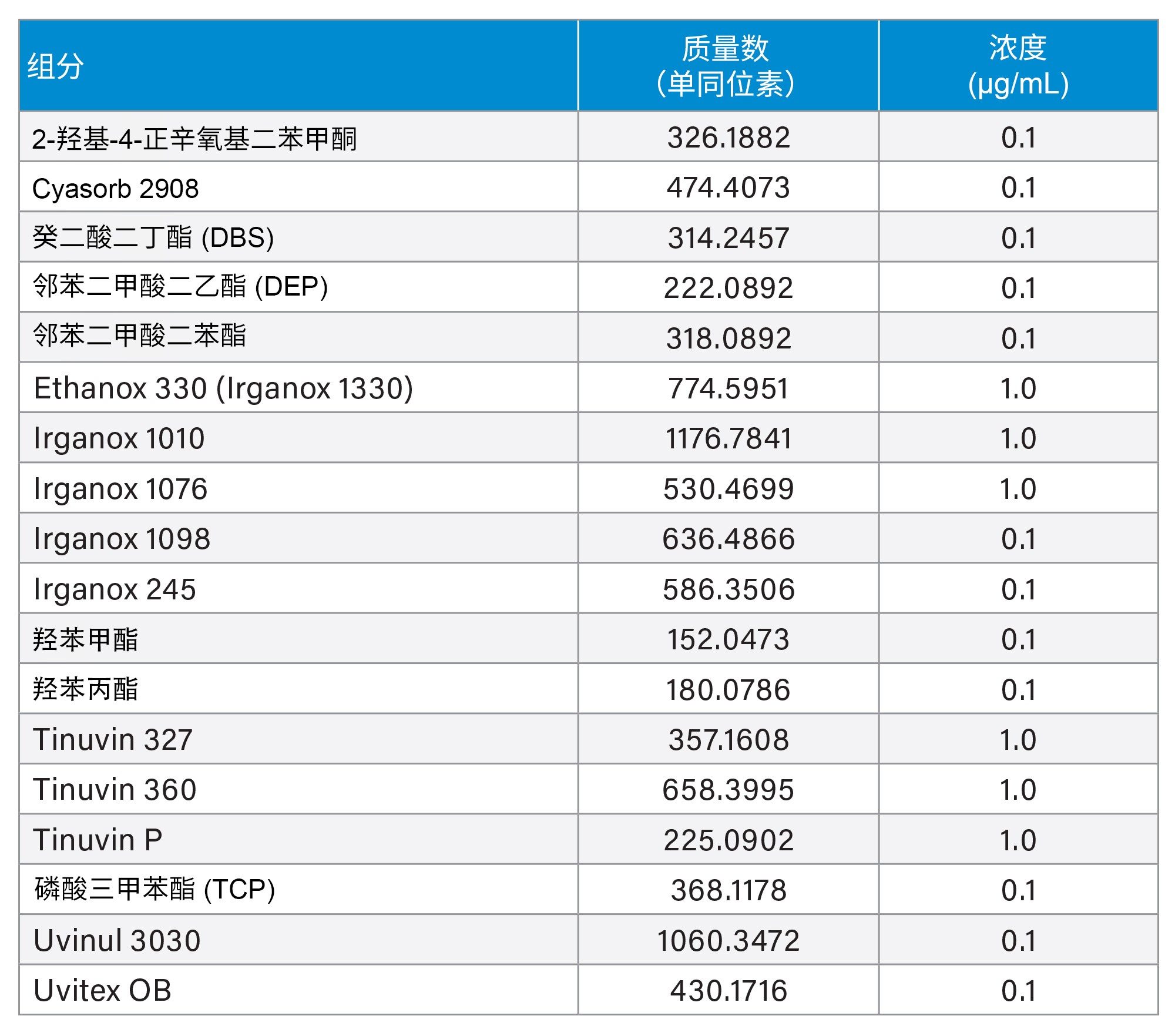
液相色谱条件
|
液相色谱系统: |
ACQUITY UPLC I-Class FTN |
|
色谱柱: |
CORTECS UPLC C18 色谱柱, 90 Å, 1.6 µm, 2.1 x 100 mm(沃特世部件号:186007095) |
|
柱温: |
40 °C |
|
进样体积: |
1 µL |
质谱条件
|
质谱系统: |
IMS QTof质谱仪 |
|
碰撞气: |
氩气 |
|
电离模式: |
ESI+,ESI- |
|
采集模式: |
HDMSE |
|
采集范围: |
50-1500 m/z |
|
扫描时间: |
0.2 s |
|
离子源温度: |
120 °C |
|
脱溶剂气温度: |
500 °C |
|
脱溶剂气流速: |
800 L/h |
|
锥孔气流速: |
50 L/h |
|
参比质量数: |
亮氨酸脑啡肽[M+H]+ m/z 556.2766 亮氨酸脑啡肽[M-H]- m/z 554.2620 |
|
毛细管电压: |
ESI+/ESI- 1 kV |
|
碰撞能量: |
ESI+ 低能量:6 eV 高能量梯度:由20 eV增加至40 eV ESI- 低能量:6 eV 高能量梯度:由30 eV增加至70 eV |
数据管理
|
色谱软件: |
UNIFI科学信息系统1.9.4版 |
|
质谱软件: |
UNIFI科学信息系统1.9.4版 |
|
信息学软件: |
UNIFI科学信息系统1.9.4版 |
方法
评估和比较三种流动相组合和三种梯度:
流动相
|
流动相组合1 (MPC 1) |
|
|
流动相A: |
水 + 0.1%甲酸 |
|
流动相B: |
甲醇 + 0.1%甲酸 |
|
流动相组合2 (MPC 2) |
|
|
流动相A: |
1 mM乙酸铵水溶液 + 0.1%乙酸 |
|
流动相B: |
甲醇 |
|
流动相组合3 (MPC 3) |
|
|
流动相A: |
水+ 1 mM醋酸铵+ 0.1%甲酸 |
|
流动相B: |
甲醇+ 1 mM醋酸铵+ 0.1%甲酸 |
结果与讨论
梯度比较
在所有流动相组合下对三种不同的梯度条件进行了评估。在不同流动相中观察到相似的趋势,因此本文重点介绍了流动相组合2 (MPC 2)和正离子模式下的结果。
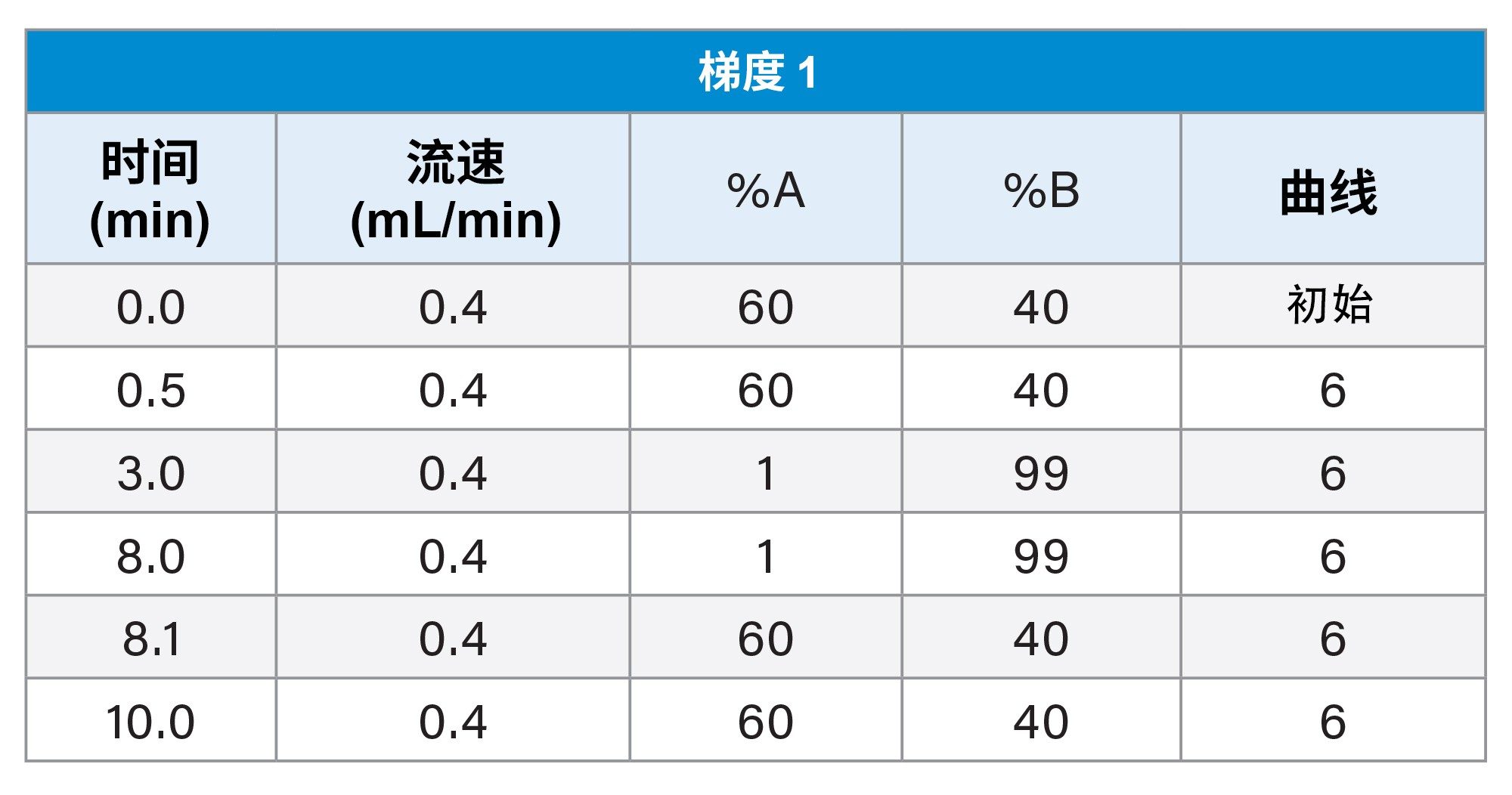
1. 梯度1
此处重点介绍LC-MS混合物(图1)和E&L混合物(图2)在梯度1和MPC 2下的LC色谱图。对于LC-MS混合物,极性分析物很早就从色谱柱中洗脱出来,这样可能会无法检测到在空体积下洗脱的某些分析物。另外,这些早期洗脱分析物的分离情况也很不理想。对于E&L混合物,即使观察到峰形良好,许多分析物也会在4分钟的时间窗内洗脱,若是复杂混合物,这可能会起反作用。
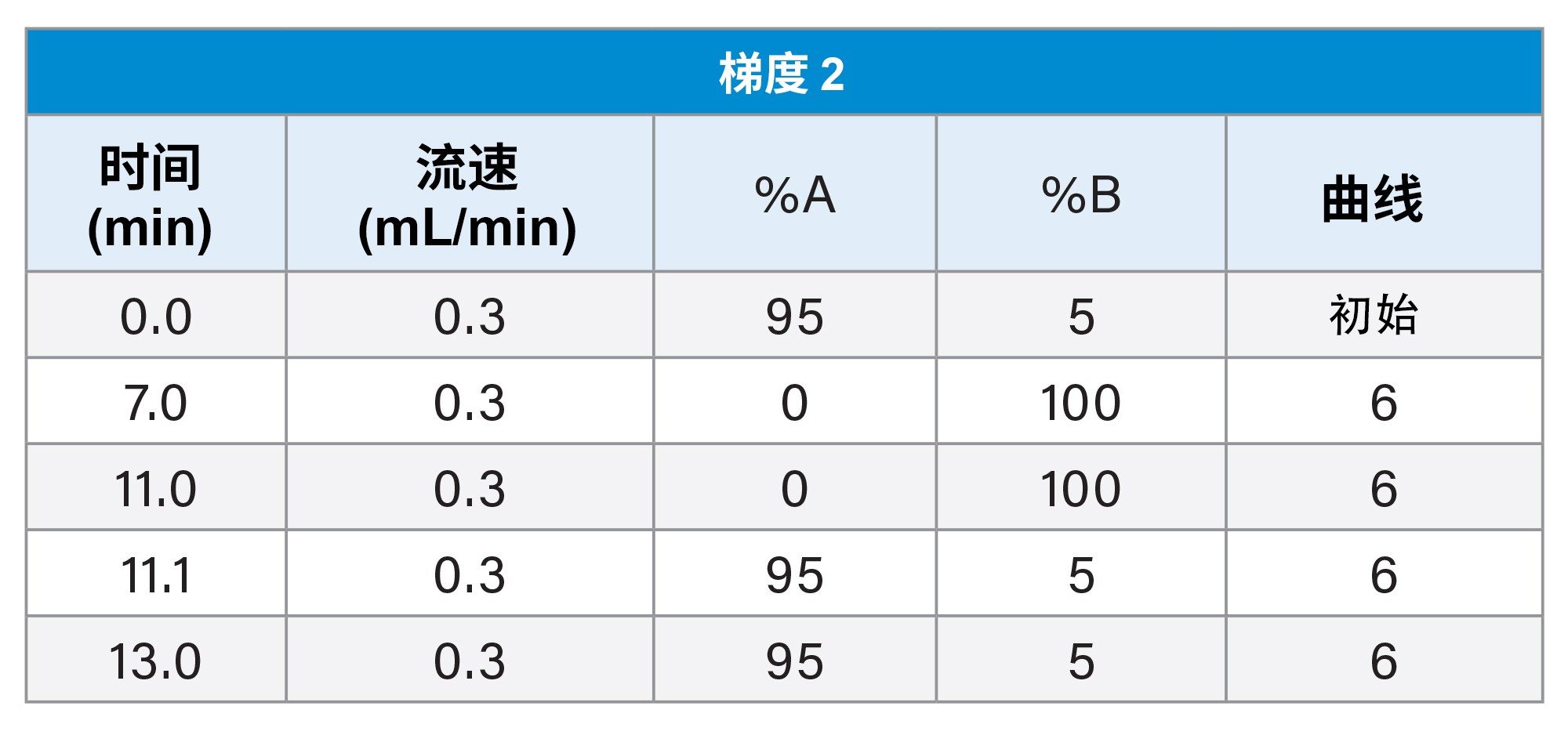
2. 梯度2
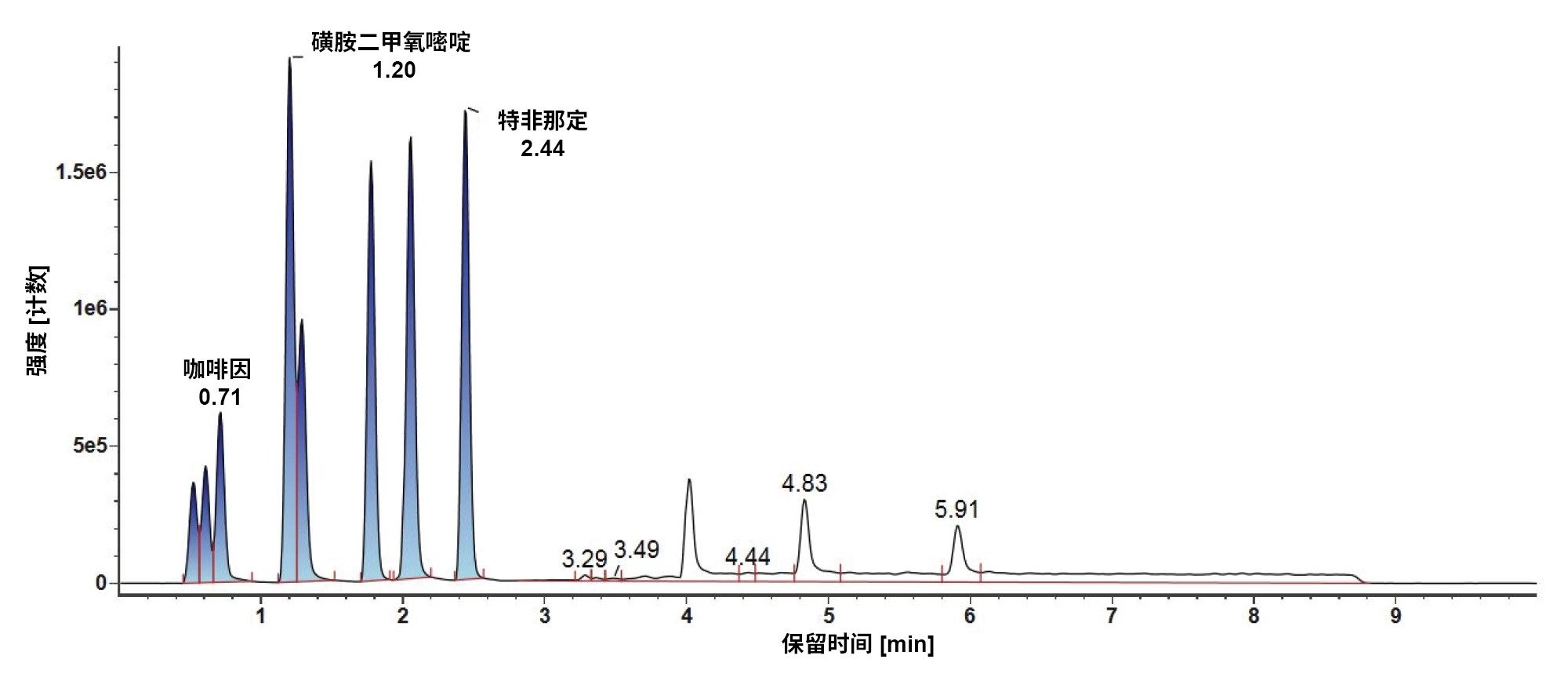
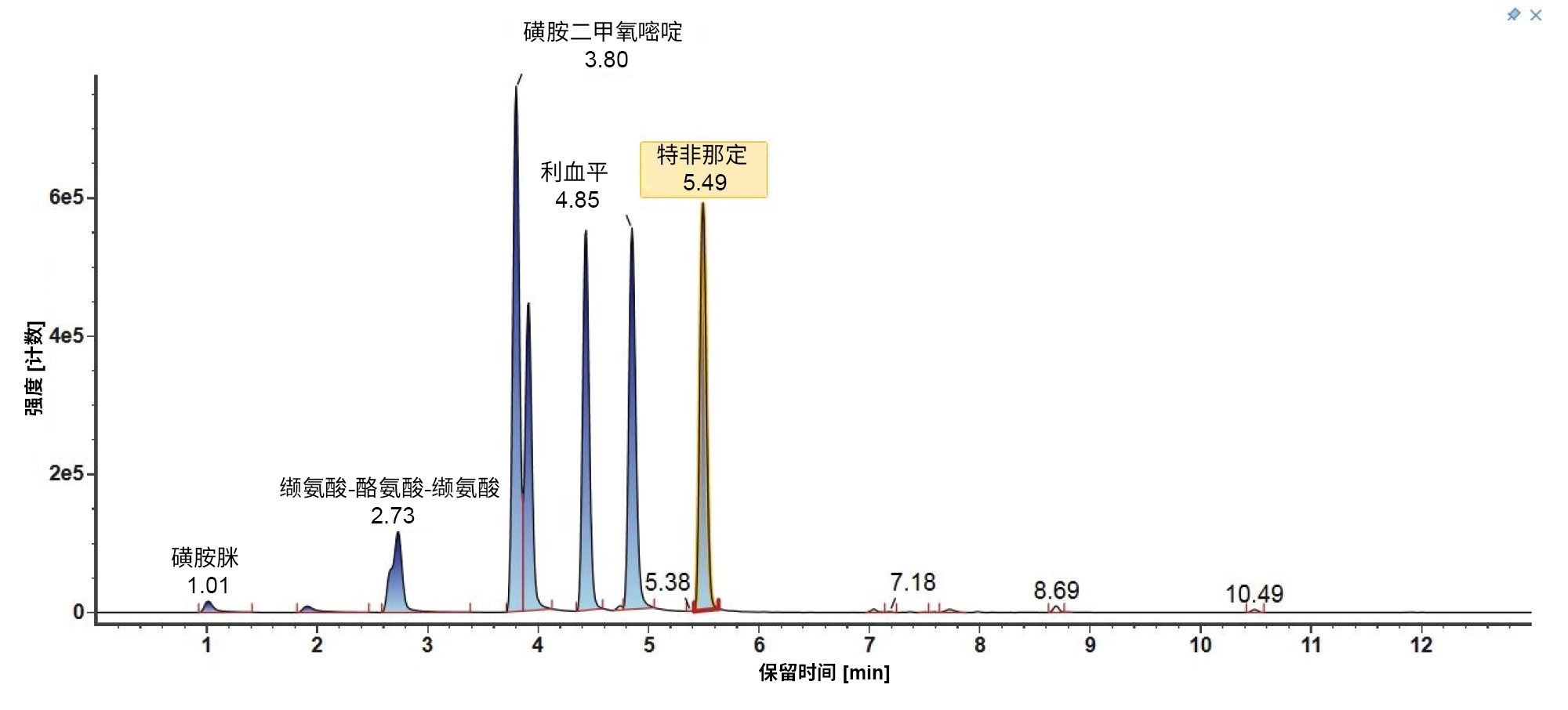
此处重点介绍LC-MS混合物(图3)和E&L混合物(图4)在梯度2和MPC 2下的LC色谱图。对于LC-MS混合物,此时极性比较强的分析物从第一分钟开始洗脱,并且这些分析物的分离效果比较好。由于该梯度的起始有机流动相百分比很低,因此可覆盖更多化学物质类型,更适用于初始通用筛查方法。对于E&L混合物,许多分析物在三分钟内洗脱,这会影响分离,而在复杂混合物分析中可能会由于竞争电荷而影响对某些分析物的检测。
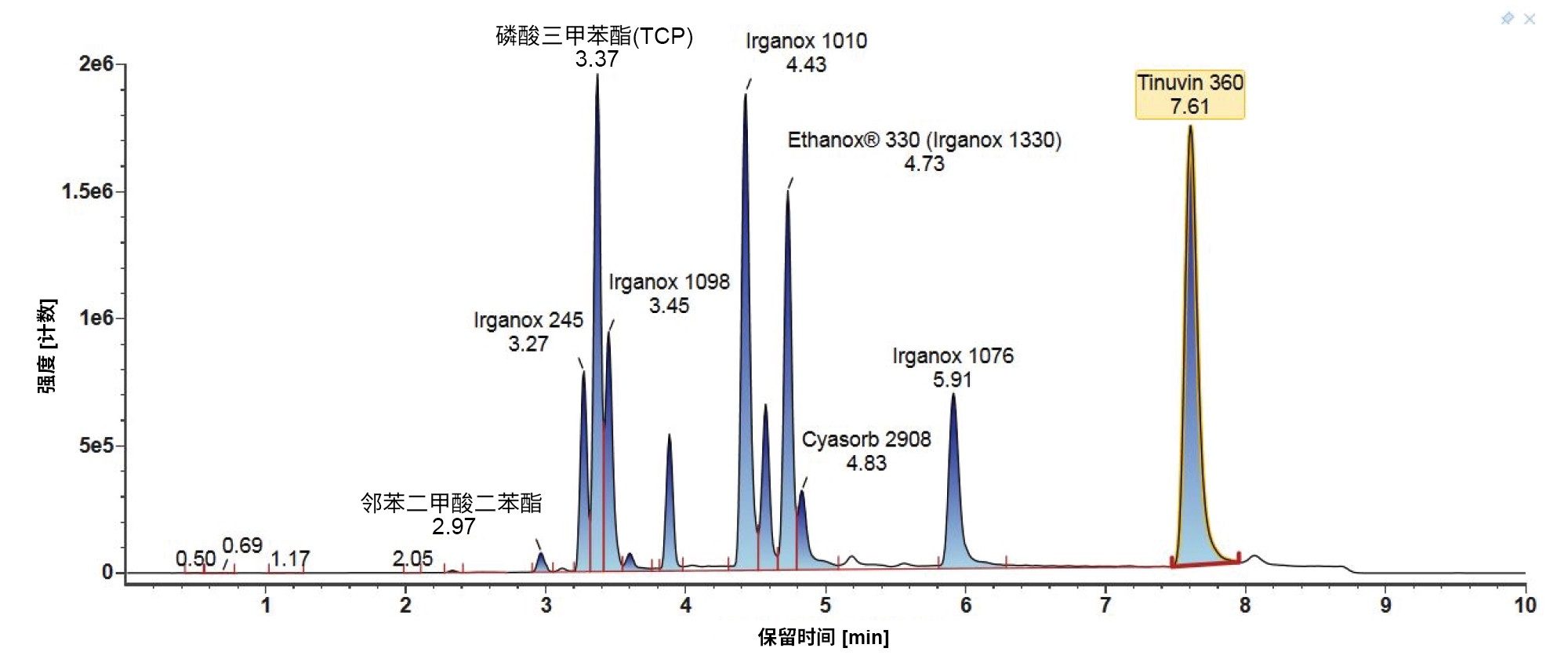
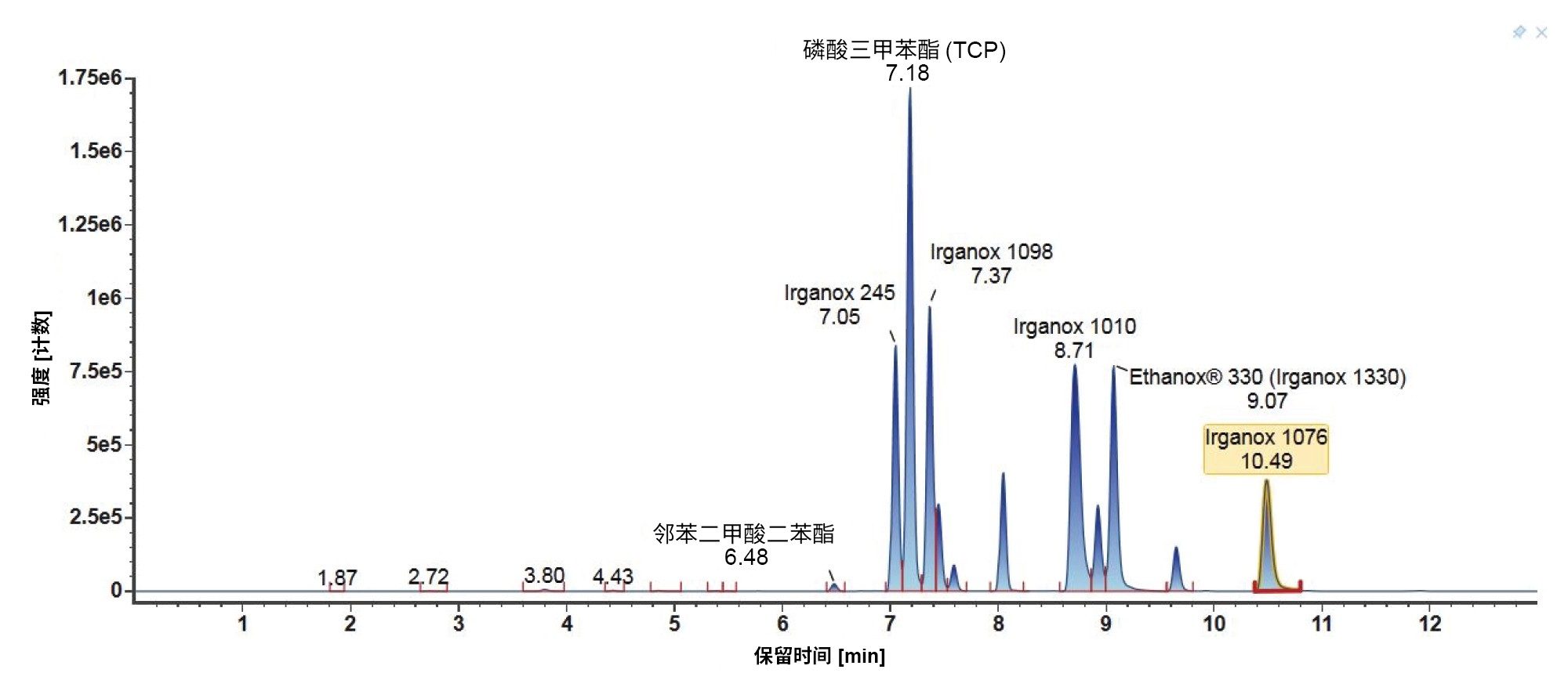
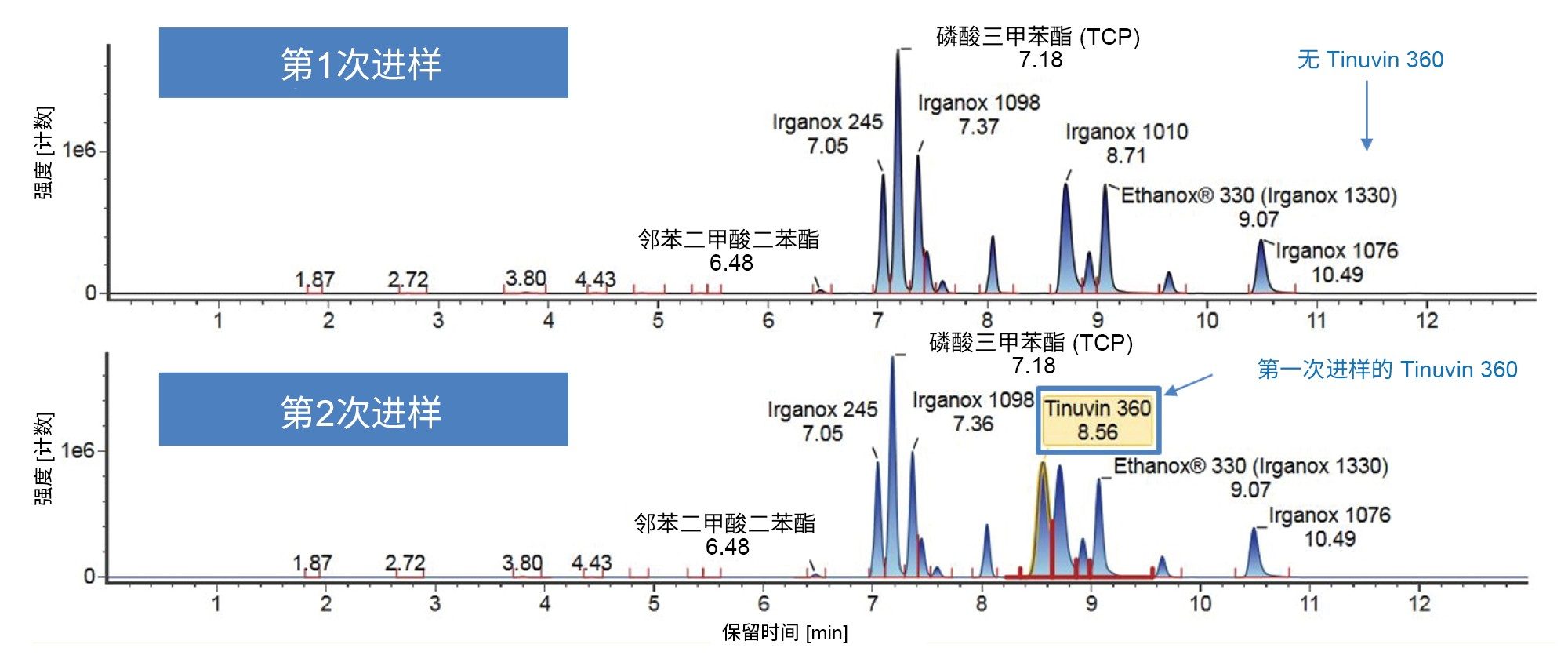
需要考虑非极性分析物,下面以Tinuvin 360为例进行了相关描述(图4)。在E&L混合物中,根据极性,Tinuvin 360应该是最后洗脱的化合物,但是,观察发现Irganox 1076是最后洗脱的分析物,根本没有观察到Tinuvin 360。出现这种情况的一个可能原因是,该梯度中的有机物保留时间太短,分析物(Tinuvin 360)没有时间从色谱柱上洗脱下来。E&L混合物的后续进样证明了这一点,即Tinuvin 360在第二次进样中比预期更早地被洗脱(图5)。这是在方法开发过程中需要注意的一个重要问题,因为这种情况不仅可能导致遗漏分析物,而且还可能导致针对靶向筛查记录的保留时间不正确,从而影响对E&L化合物的鉴定。
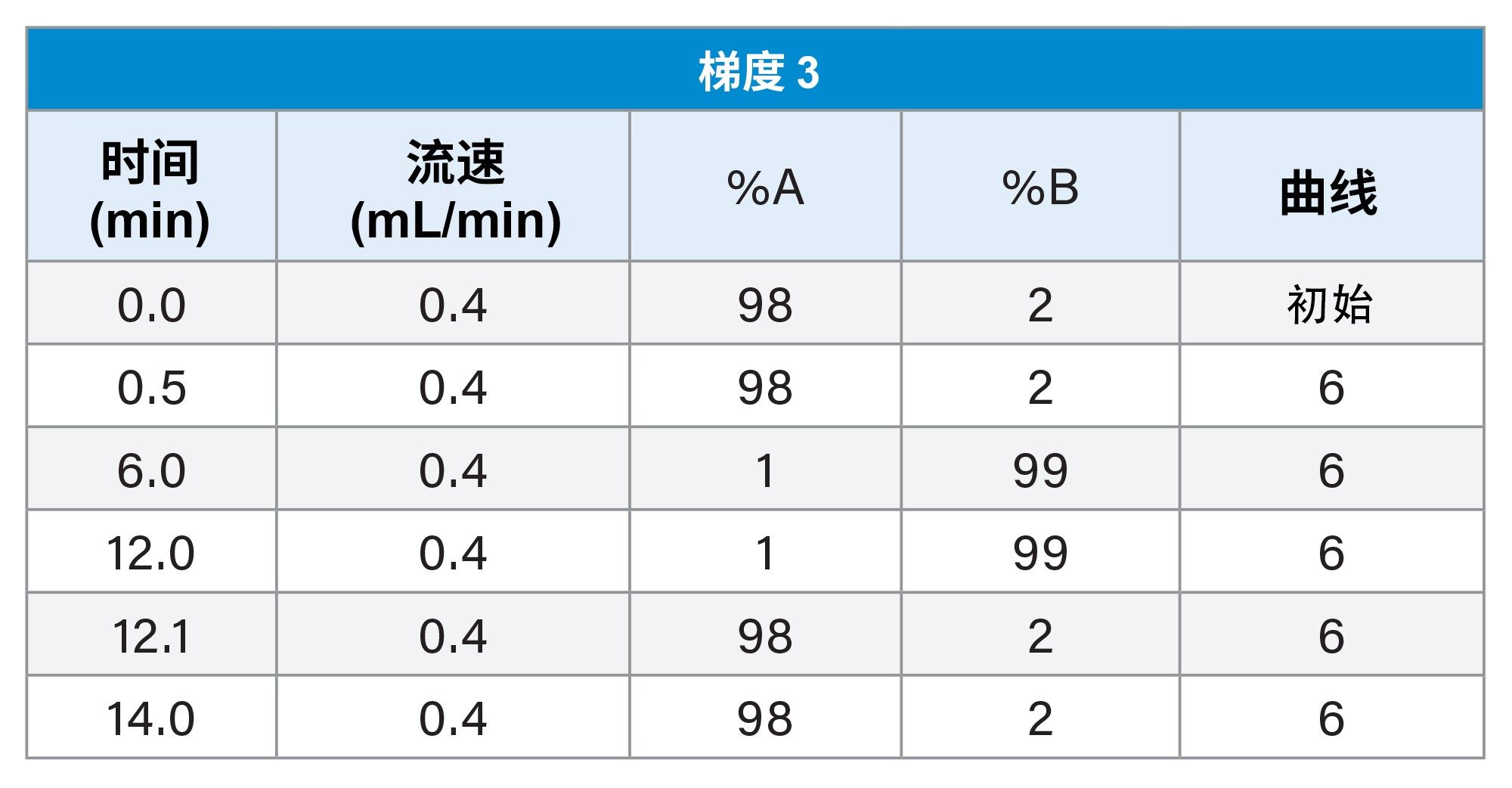
3. 梯度3
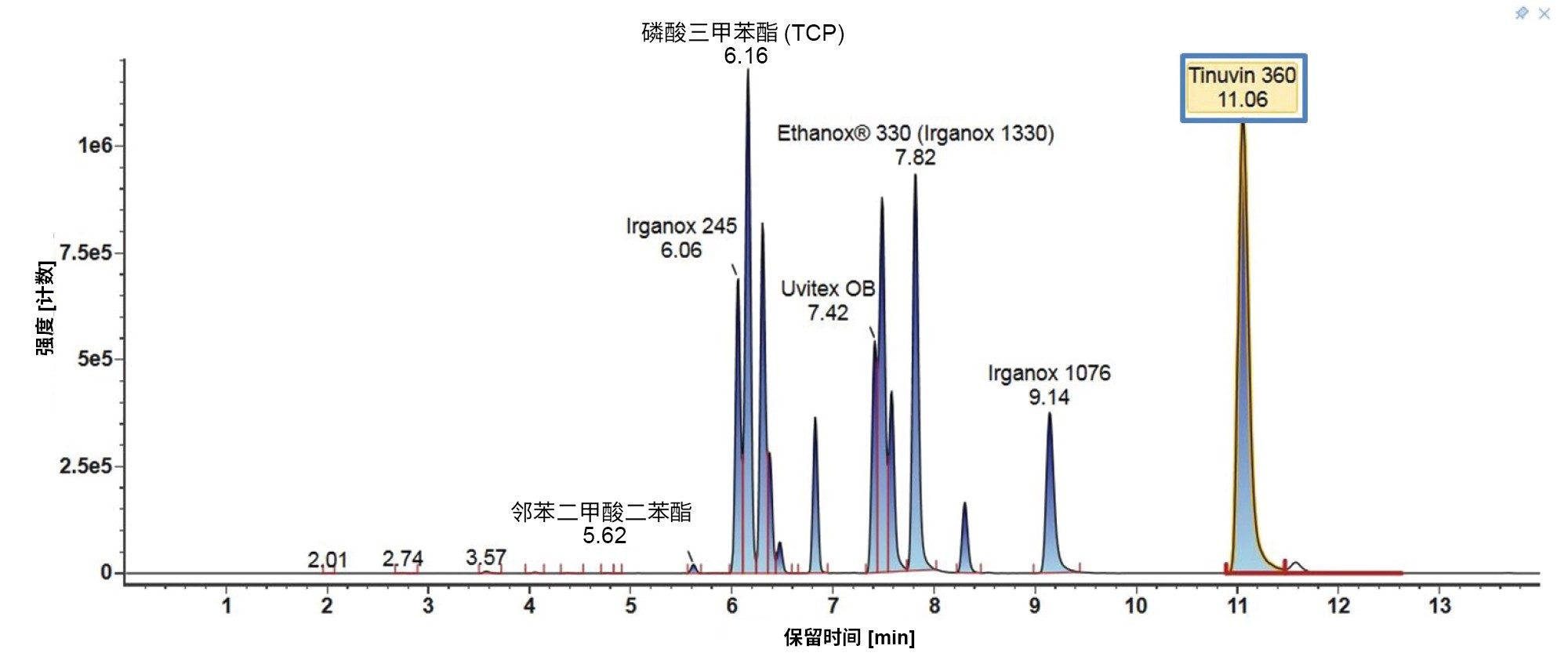
此处重点介绍LC-MS混合物(图6)和E&L混合物(图7)在梯度3和MPC 2下的LC色谱图。对于LC-MS混合物,与梯度2类似,起始有机流动相百分比较低,因此极性比较强的分析物在色谱柱上保留得比较好,观察发现其分离效果也比较好。对于E&L混合物,大多数分析物此时的洗脱时间窗较宽,并且分离效果比较好。有机物保留时间很长,可确保Tinuvin 360是所有进样中最后洗脱的化合物。
4. 梯度比较总结
通过这些实验,我们可以得出结论,在开发通用E&L筛查方法中,确保梯度起始有机流动相百分较低以及梯度结束时有机流动相保留时间足够长对于检测多种分析物非常重要。在梯度3中综合考虑了这些因素,因此所有化合物的分离效果良好,并且降低了分析物遗漏(无论是在空体积下还是在色谱柱上保留时)风险。
流动相比较
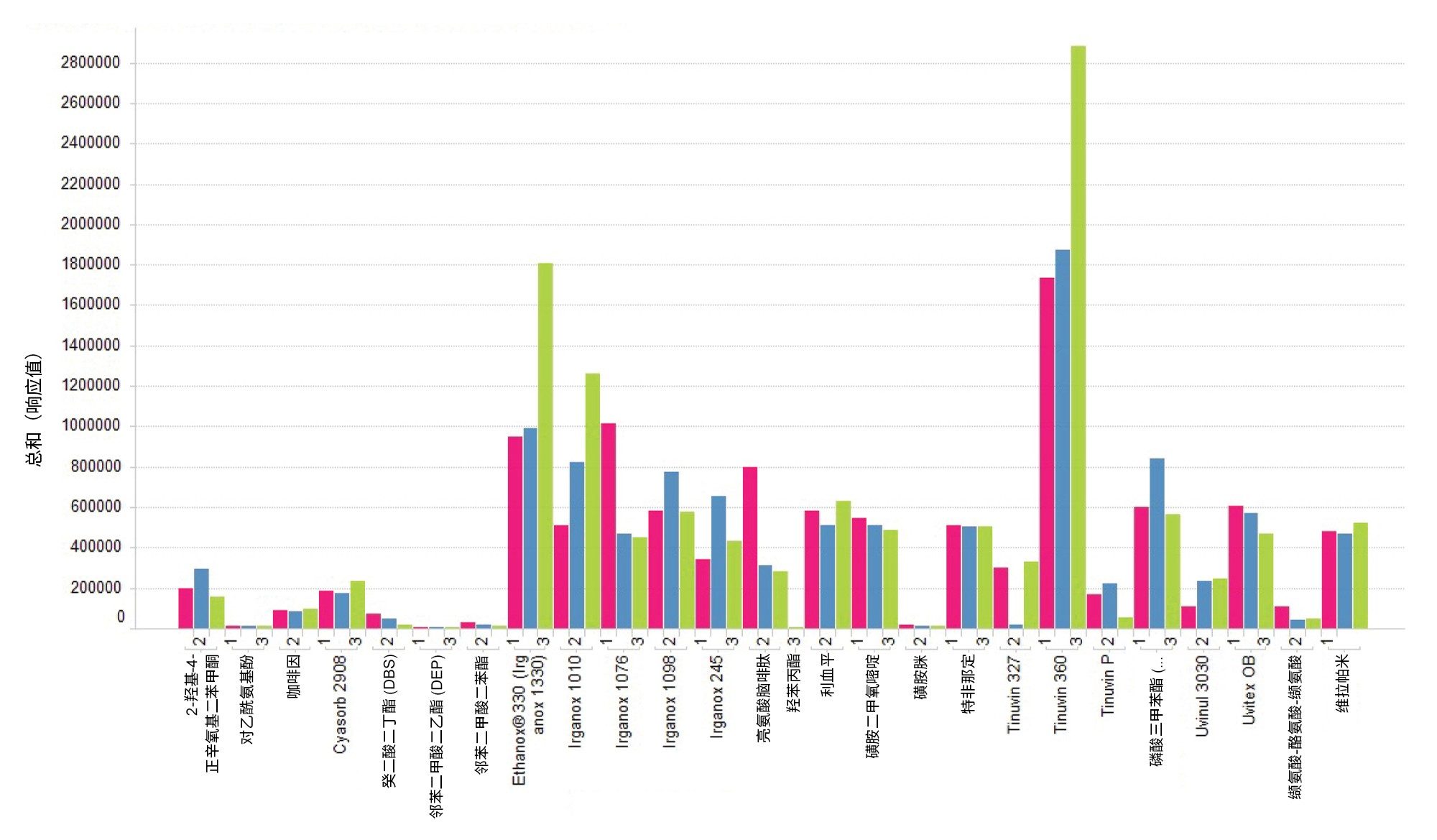
在三种不同梯度下对三种不同的流动相组合进行评估。大多数分析物在不同梯度下显示的结果相似,因此本文重点讲述梯度3下的结果。结果如图表所示,该图表显示了每种流动相组合下每种分析物三次进样的平均MS(质谱)响应。流动相组合1 (MPC 1)以粉色突出显示,流动相组合2 (MPC 2)以蓝色突出显示,流动相组合3 (MPC 3)以绿色突出显示。
1.正离子模式
图8显示的是在实验部分所述三种流动相组合下LC-MS和E&L混合物中所有分析物的正离子响应图。大多数分析物在不同流动相组合下出现轻微变化,并且一部分E&L混合分析物在正离子模式和MPC 2或MPC 3下响应会增加。
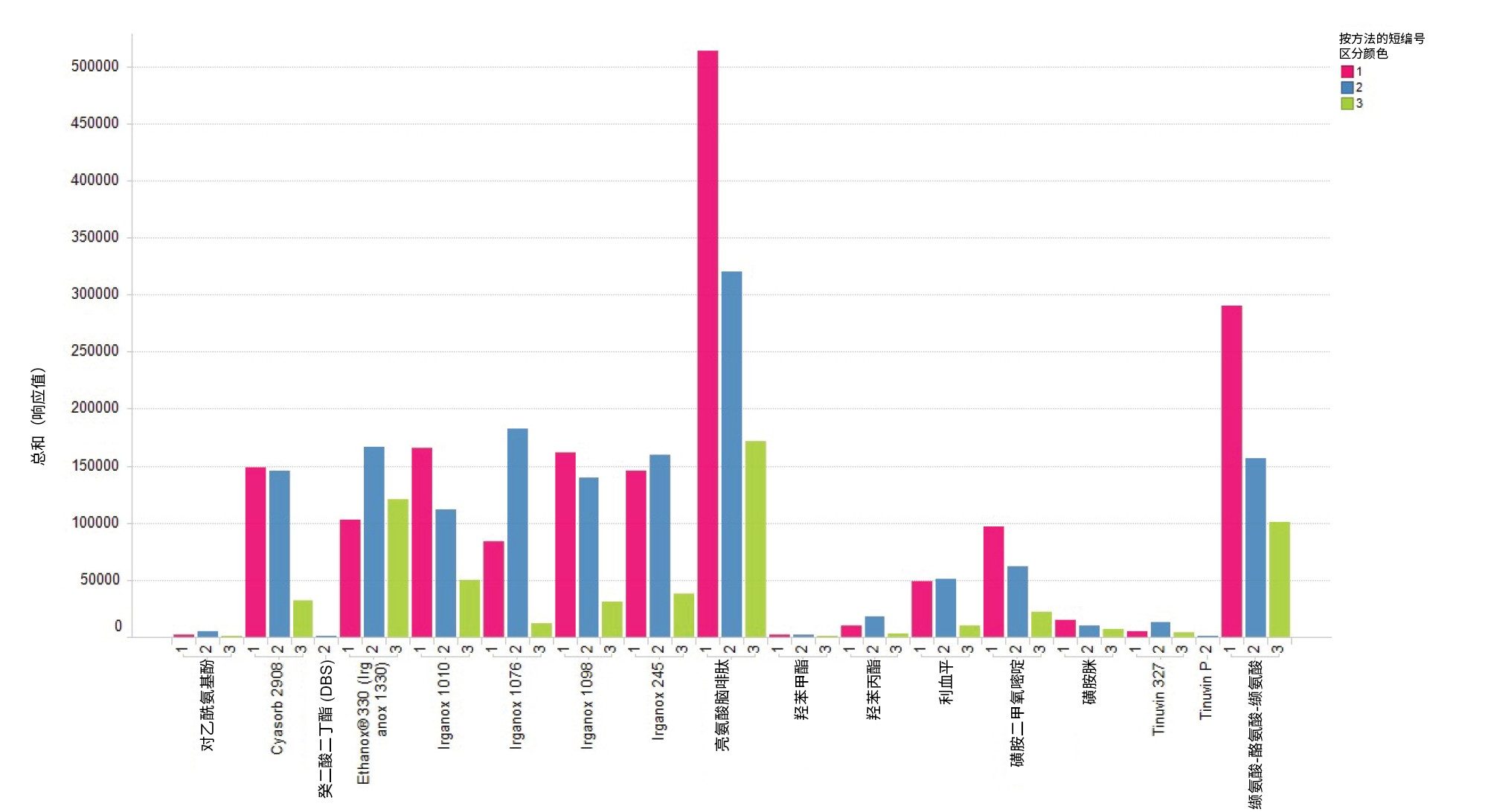
2. 负离子模式
图9显示的是负离子模式下LC-MS混合物和E&L混合物分析物的流动相组合。与MPC 3相比,MPC 1和MPC 2在负离子模式下使所有分析物的响应增加。
3. 流动相比较总结
流动相比较表明,流动相组合对分析物电离具有影响。在设计初始通用筛查方法时,需要寻找不会明显偏重某些分析物类型的添加剂。从上述数据来看,在正离子和负离子模式下使用流动相组合2时大多数分析物可得到最佳平均响应。
图8和9显示了每种分析物中观察到的所有加合物的综合响应,如图所示,因为在流动相中使用添加剂时,一部分分析物的响应可能会增加,因此重点是考虑萃取物电离中的加合物形成。使用含有乙酸铵等添加剂的流动相时,形成铵加合物可能会很有利,具体视萃取物化学结构而定。
在流动相中添加乙酸铵等添加剂可能会形成不同类型的加合物([M+NH4]+、[M+Na]+),必须将这一点纳入分析物总响应之中,因为它的强度可能比质子化物更大。
其他注意事项
经过研究和比较,发现梯度3和流动相组合2能够针对所有分析物和不同离子模式在实现良好色谱分离效果与获得良好MS响应之间达到理想平衡点。本文还阐述了关于进一步改善通用E&L筛查应用方法的其他一些注意事项。
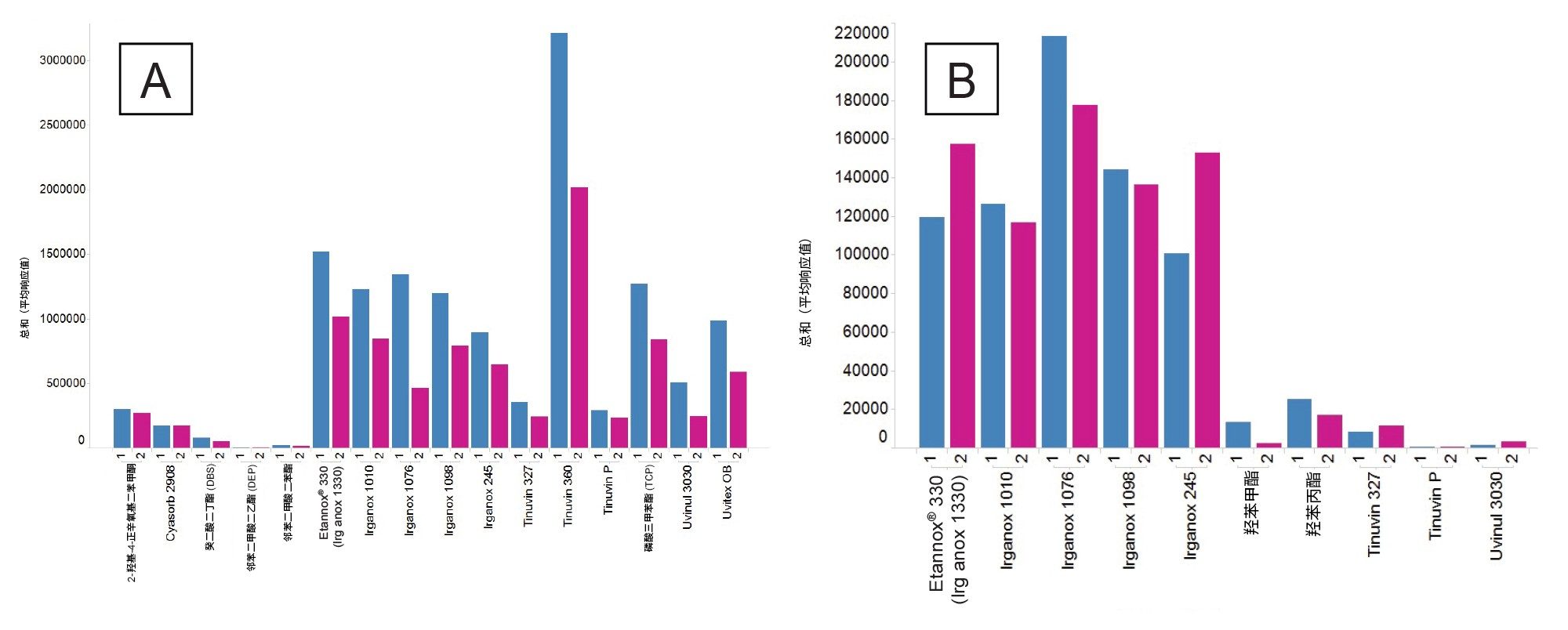
1. 溶剂评估
在执行E&L筛查研究时,需要考虑溶剂和添加剂的质量,以减少LC色谱中的基质背景。此外,还需要评估添加剂的强度。甲酸的酸性比乙酸强,更有利于电离(乙酸pKa约为4.8,甲酸pKa约为3.8,在酸浓度相同的流动相中,使用甲酸时,pH值较低),使用E&L混合物进一步评估后发现,在MPC 2下将其作为替代添加剂添加到流动相A中时,大多数E&L分析物混合物的响应略有增加(图10)。
2. 压力变化
通常,E&L筛查方法可能需要在不同LC系统(压力限制可能有所不同)之间转移。由于所用有机流动相是甲醇(粘度比乙腈高),为了避免压力限制较小的系统承受的压力过大,降低了流速,并提高了柱温,以减轻这种影响。通过降低流速,进一步延长了有机物保留时间,从而可以解释这种变化。
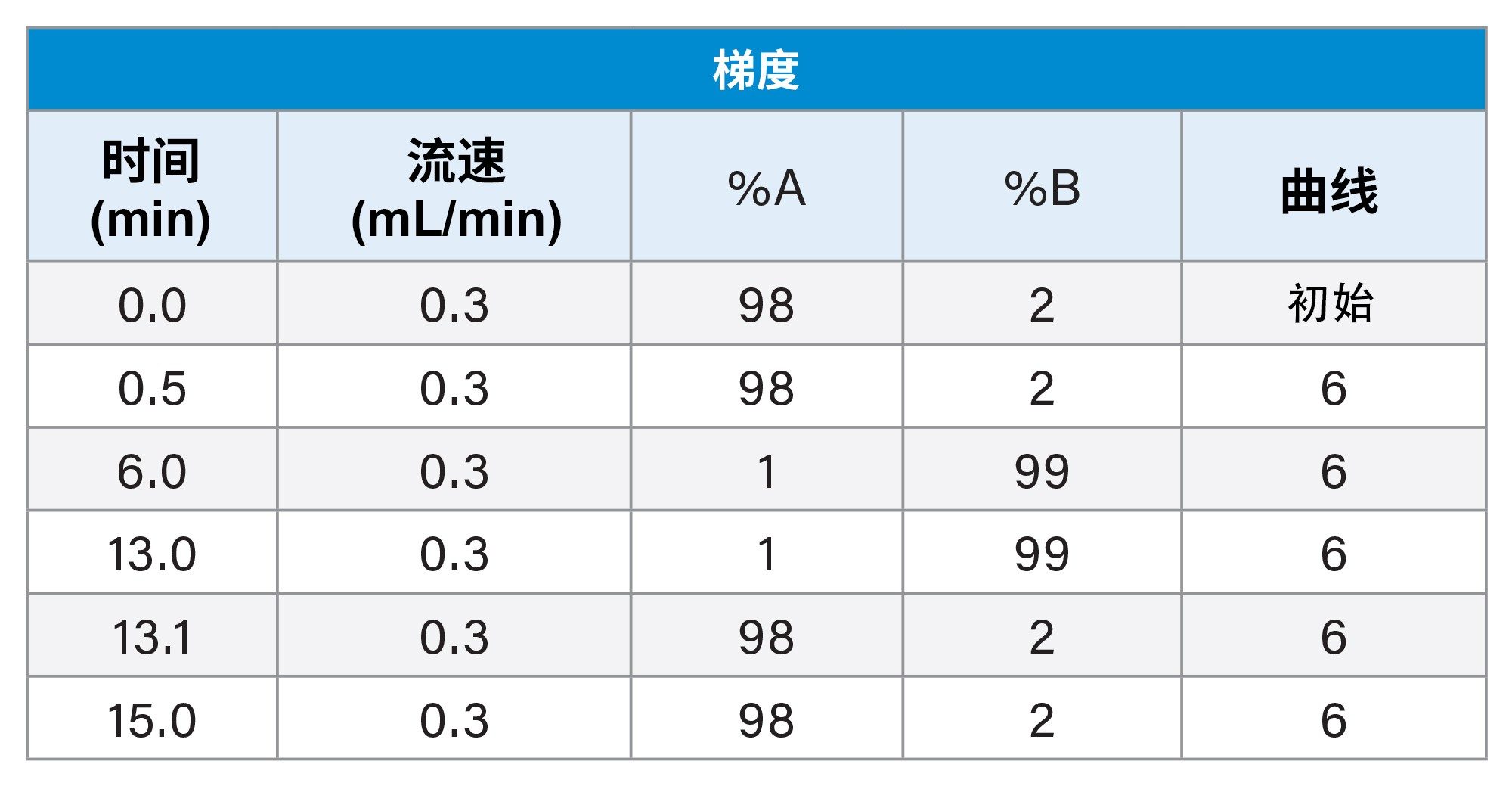
3. 最终方法条件
在评估现有LC-MS筛查方法以及其他一些考虑因素后,针对内部E&L LC-HRMS筛查确定了最终通用筛查条件,下面对此作了详细描述。
UPLC条件
|
色谱柱: |
Waters CORTECS UPLC C18色谱柱, 90 Å, 1.6 µm, 2.1 x 100 mm(部件号:186007095) |
|
柱温: |
50 °C |
|
进样体积: |
1 µL |
流动相组合
|
流动相组合 |
|
|
流动相A: |
水+ 1 mM醋酸铵+ 0.1%甲酸 |
|
流动相B: |
甲醇 |
梯度
结论
本研究评估了初始首程通用E&L筛查方法。需要注意的是,所述方法是基于多种现有方法开发的,本研究的目的并不是基于基本原理创建分析方法。通过全面研究和比较,评估了不同条件对不同类型化学物质的影响。此外,本研究还通过纳入进一步考虑以创建单一通用筛查方法,为E&L分析人员提供支持。
通过所述实验开发的方法能够大幅降低最后洗脱化合物的残留风险,并且能够提高早期保留化合物的色谱分离度。通过作出其他改变,可以在压力限制不同的LC系统之间实现方法转移,同时将溶剂强度纳入考虑。该方法在对多种极性化学物质进行筛查时总体灵敏度出色,并且不会偏重某些类型的化学物质。
一般,在针对E&L开发和简化LC-MS筛查方法时,可以考虑以下主要观察结果:
—为通用筛查方法创建色谱梯度时,起始有机百分比和有机物保留时长对于捕获极性范围广泛的分析物非常重要,同时能够大幅降低由于在色谱柱上没有保留或保留太多而导致的分析物遗漏风险。
—选择流动相时,选择不偏重特定分析物类型的溶剂、添加剂和缓冲液的组合对于初始通用筛查方法来说是一个不错的起点。在流动相中添加缓冲液可以增强某些典型萃取物的响应,同时还能减少不太有利的加合物的形成。
综上所述,本应用纪要就如何开发初始通用E&L筛查方法提供了建议。但是,如果使用该方法筛查一组特定分析物,或者已知当前化学物质的类型,建议对方法作进一步优化。可以通过在正和负离子模式下使用不同的流动相以及添加不同的添加剂,从而开发出更有针对性的筛查方法。
参考资料
- Food and Drug Administration, 2000, Code of Federal Regulation Chapter 21.
- Official Journal of the European Union, 2011, Regulation 10/2011/EU.
- Food and Drug Administration, 2015, Analytical Procedures and Methods Validation for Drugs and Biologics Guidance for Industry https://www.fda.gov/files/drugs/published/Analytical-Procedures-and-Methods-Validation-for-Drugs-and-Biologics.pdf.
- Norwood, D.L., Paskiet, D., Ruberto, M. et al.Best Practices for Extractables and Leachables in Orally Inhaled and Nasal Drug Products: An Overview of the PQRI Recommendations.Pharm Res 25, 727–739 (2008). https://doi.org/10.1007/s11095-007-9521-z.
- ISO 10993–18:2020 Biological evaluation of medical devices–Part 18: Chemical Characterization of Medical Device Materials within a Risk Management Process, https://www.iso.org/standard/64750.html.
720007492ZH,2022年1月